




Contents
1 引言
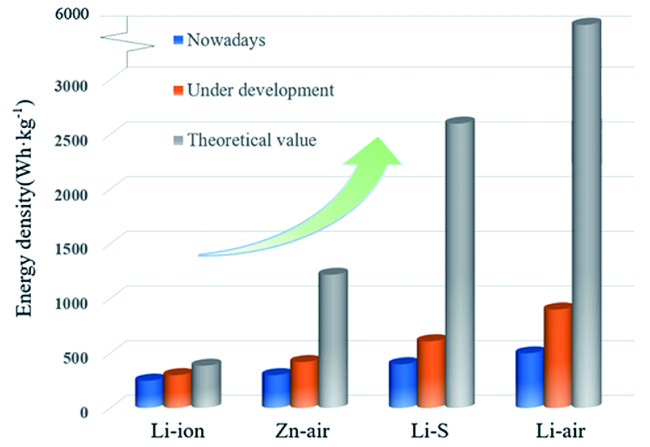
2 非水系锂空气电池及其正极材料
2.1 非水系锂空气电池及其充放电机理
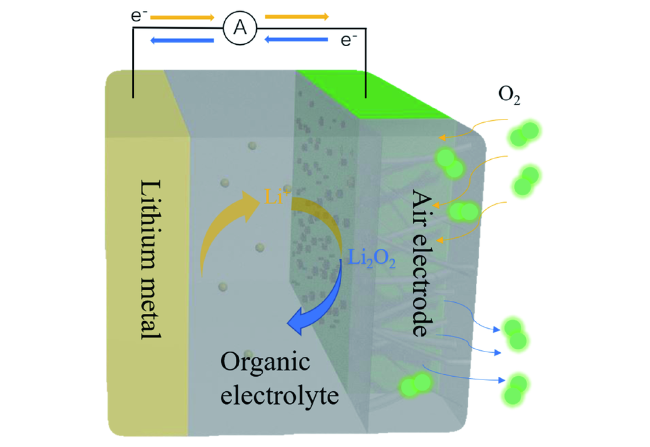
2.2 正极材料
3 碳基正极材料研究进展
3.1 基于一般多孔碳材料的正极材料

图3 (a)3DOM材料结构的模型,(b)产物在电极表面的形成与沉积过程,具有大孔径(上)和小孔径(下)的3DOM碳,(c)在电流密度100 mA·和截止容量500 mAh·条件下,不同孔径的3DOM电极循环性能测试,35 nm孔径(黑)和12 nm孔径且具有高(绿)和低(红)壁密度的3DOM碳,(d)不同孔径的3DOM电极的充放电曲线[51]Fig. 3 (a) Model for the structure of 3DOM carbon.(b) Simplified 2D representation of the formation and accumulation of byproducts on 3DOM carbon with large(top) and small pores(bottom).(c) Cycling performance of bare 3DOM carbon of different pore sizes: 3DOM carbon with 35 nm pores(black) and 12 nm pores with high(green) and low(red) wall densities. Capacity is limited to 500 mAh·, rate=100 mA·.(d) Discharge/charge curves normalized to pore volumes[51] |
3.2 基于石墨烯和类石墨烯类碳材料的正极材料
3.3 基于其他类型碳材料的正极材料
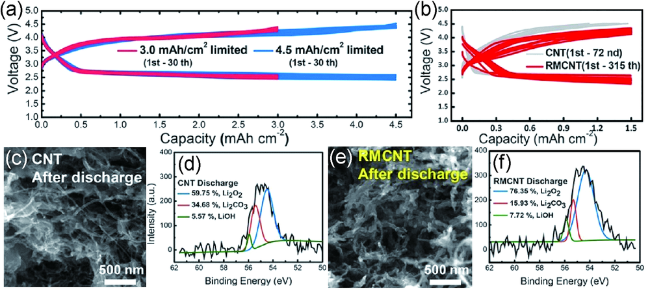
图5 (a)基于RMCNT电极的锂空气电池放电曲线,电流密度为1.5 mA·cm-2,截止容量为3.0 mAh·cm-2(紫色)和4.5 mAh·cm-2(蓝色),(b)不同催化剂材料的锂空气电池的循环性能,放电后CNT(c,d)和RMCNT(e,f)电极的SEM图像及其XPS分析[68]Fig. 5 (a) Voltage profiles of RMCNT monolith electrode at 1.5 mA·cm-2 with cutoff capacities of 3.0 mAh·cm-2(purple) and 4.5 mAh·cm-2(blue).(b) Voltage profiles of CNT monolith electrode and RMCNT monolith electrode. SEM images of CNT(c),and RMCNT(e) electrodes after discharging. Chemical analysis of the electrode after discharging by XPS: CNT(d) and of RMCNT(f)[68] |
4 碳基空气正极的性能提升研究
4.1 掺杂对于碳基正极材料性能影响的研究
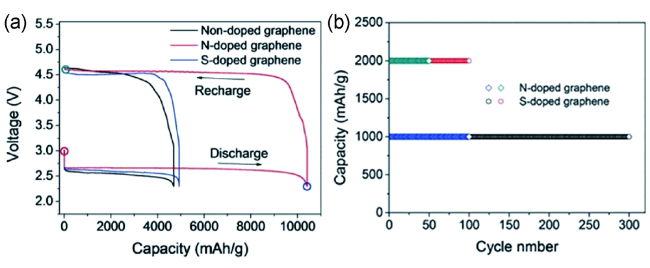
图6 (a) 电流密度200 mA·g-1,电压范围2.3~4.6 V,不同石墨烯电极的电池充放电曲线,(b)N,S掺杂石墨烯电极的电池循环性能测试[73]Fig. 6 (a)Discharge-charge profiles of Li-O2 cells using the nanoporous graphene electrode(the cells were tested at 2.3~4.6 V with a current density of 200 mAh·g-1).(b) Cycling stability of the nanoporous N- and S-doped graphene-based Li-O2 cells[73] |
4.2 碳基正极材料OER催化性能的提升研究
4.3 碳基空气正极的结构设计研究

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图7 (a)Si基底上生长的VA-NCCF的SEM图像,(b)单个VA-NCCF的TEM图像,(c)VA-NCCF电极表面Li2O2沉积状态概念图,(d)VA-NCCF为电极的锂空气电池倍率性能图,(e)放电产物的TEM图像(红色箭头指向纺锤形Li2O2,蓝色箭头指向球形Li2O2)[84]Fig. 7 (a) SEM image of a VA-NCCF array grown on a piece of Si wafer.(b) TEM image of an individual VA-NCCF.(c) The sketch of Li2O2 grown on a coral-like carbon fiber.(d) Rate performance of the VA-NCCF electrode.(e) TEM image of a discharged catalyst fiber(blue and red arrows indicate spherical- and spindle-shaped Li2O2 particles, respectively[84] |