



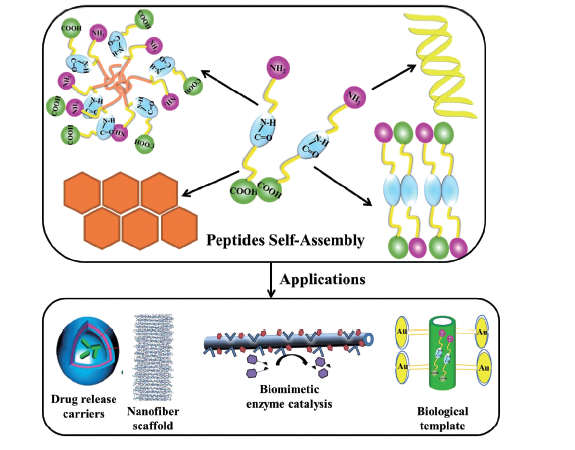
Contents
1 Introduction
2 Mechanism of peptides self-assembly
3 Research progress in self-assembly of stimulus-responsive peptides
3.1 pH-responsive
3.2 Temperature-responsive
3.3 Solvent-responsive
3.4 Light-responsive
3.5 Ultrasound-responsive
3.6 Ion-responsive
4 Application of peptides self-assembly
4.1 Drug carriers and release
4.2 Tissue scaffolds promote wound healing
4.3 Repair of spinal cord injury
4.4 Biomimetic-enzyme catalysis
4.5 Manufacturing optical materials
4.6 Analog photosynthesis system
4.7 Synthetic template
5 Conclusion and outlook
1 引言
自组装作为超分子化学的核心在自然界中随处可见。自组装是指分子在没有外力介入的情况下,通过分子间的非共价键作用力[1](如疏水作用、范德华力、氢键、π-π堆积作用等)聚集成有序的结构体(如球形胶束、囊泡、线状、带状、层状、柱状、管状、球状和网状等结构[2,3,4,5,6])的过程[7]。最终,所获得的自组装体能呈现出组装单元本身不具备的某些特性,如光、电、生物特性等[8]。近年来,分子自组装在构筑新型功能材料中表现出了巨大的潜力,成为备受关注的研究领域[9,10,11]。随着科学工作者的不断努力研究,分子自组装已经为化学、材料、生物医药、物理、制造、纳米科学等学科提供了新的机会[12,13,14,15,16,17]。
肽分子是一种由两个或多个氨基酸序列构成的常用组装基元。由于其与组织、细胞等生物成分有良好的相容性[18],使得肽可以被人体吸收利用。另外,肽分子还具有较好的可修饰性[19,20],研究人员通过对其侧链基团种类、结构的调控,可使其广泛应用于生物医疗领域[21,22,23,24,25,26,27]。肽对pH、温度、金属离子等环境因素的敏感性使其在自组装生物材料方面拥有良好的可控性和生物智能响应性[28,29,30,31,32,33,34]。因此,人们对肽自组装的兴趣逐渐增加[35,36,37,38]。1993年Ghadiri等设计用八肽自组装成纳米管[39],之后,Zhang等[40]在美国科学院院报上报道了一种包含16个氨基酸的离子互补型多肽,该多肽通过离子互补相互作用自组装成纳米纤维从而形成水凝胶。自此,多种多样的自组装的多肽开始进入人们的视野。目前已经报道了二肽[41]、模型堆积多肽[42,43]、两亲多肽[5]等多种肽自组装单体。这些多肽单体能够组装成多种高度有序的纳米或微米级结构,如纳米纤维、纳米管、囊泡、纳米粒等[44]。肽自组装体显示出了不同的力学性质[45,46]、电化学性质[47]、电学性质[48,49]、光化学性质[50,51]、热力学稳定性[52]和环境相容性[19],可以应用于纳米科技、能源、电子学、力学等多个领域且有高效和大规模发展的潜能[53]。
本文详细介绍了肽自组装的机理以及刺激响应型肽自组装在生物医药、材料、合成模板等方面的应用和研究进展,总结了肽自组装结构稳定、生物相容性良好、可生物降解等优点。最后,基于目前肽自组装存在的一些问题(如影响肽自组装结构的外界因素不易精准把控、自组装的研究与生命科学领域的交叉程度低等)对肽自组装的发展做了展望。
2 肽自组装机理

氢键作为构建生物体系组织结构的重要驱动力,在肽自组装形成稳定结构体系的过程中发挥着重要作用。肽链中含有的氨基和羧基两个特征基团以及肽键自身的酰胺基团为氢键的形成提供了丰富的位点。氢键的选择性和高度方向性可以诱导肽转化成为不同的一维、二维和三维纳米结构体系,如图1(a)所示。
但是,氢键的形成极易受溶液的极性和pH的影响,改变溶液的pH会使肽链中的氨基、羧基或其他侧链基团发生质子化或去质子化,从而形成带正负电荷的氨基酸肽。带正负电荷的氨基酸肽(如带正电荷的精氨酸、赖氨酸、组氨酸和较为特殊的鸟氨酸以及带有负电荷的天冬氨酸和谷氨酸缩合成的肽)又会产生静电作用,并在静电作用的驱动下产生离子键作用或与水分子发生电荷-偶极作用,这些作用有利于肽自组装结构的稳定性,如图1(b)所示。
同时,在生物体中,由于水分子的存在,氢键往往也会伴随着疏水作用一起影响肽分子的自组装。疏水作用是肽自组装的另一种重要的驱动力。其本质是疏水基团为了避免与水分子接触而被迫相互靠近聚集而产生的作用力,不具有方向性。含脂肪族烷基侧链的氨基酸缩合成的肽的自组装就是由疏水作用驱动的,如含丙氨酸、亮氨酸、甲硫氨酸的肽等。此类氨基酸残基一般包埋于组装体内部,形成稳定的疏水性内核,如图1(c)所示。
还有一类含芳香环类的肽分子则是由π-π堆积作用驱动自组装的,如含有苯丙氨酸、色氨酸的肽分子,如图1(d)所示。
3 刺激响应型肽自组装研究进展
3.1 pH响应型肽自组装
pH是一个易控制且应用非常广泛的外部刺激,因此,pH响应型肽自组装是目前研究最为广泛的一类刺激响应型肽自组装。溶液pH的改变可以引起氢键的供体和受体位点之间的竞争性溶剂化,进而破坏氢键的平衡和引起静电排斥,最终可以控制肽自组装成不同形态的功能分子[58]。pH主要作用于肽序列的特征基团氨基、羧基以及链上的氨基酸残基。当环境中pH值低于肽链中氨基酸的pKa值时,肽链中的羧基不发生电离,氨基发生质子化,有利于分子间氢键的形成,使肽在酸性溶液中聚集而沉降。当pH值高于pKa值时,多肽链上的羧基发生去质子化,减弱了肽分子间的氢键,此时继续升高pH值,会使肽链中的其他基团也发生去质子化,进而削弱了分子间的盐桥作用,静电作用也开始减弱,肽链重新聚集发生重排,如下式所示。
pH<pKa —NH2 + H+→—N
pH>pKa —COOH-H+→—COO-
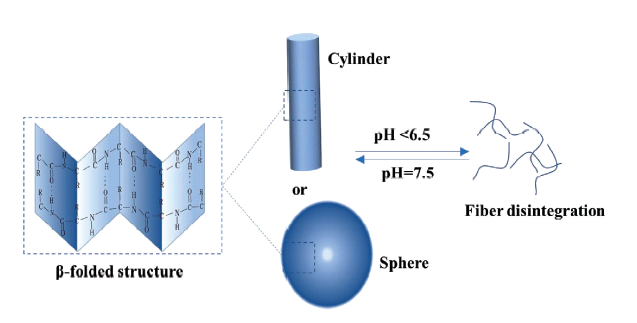
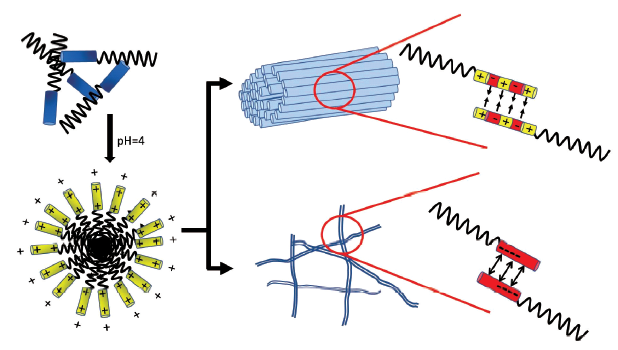
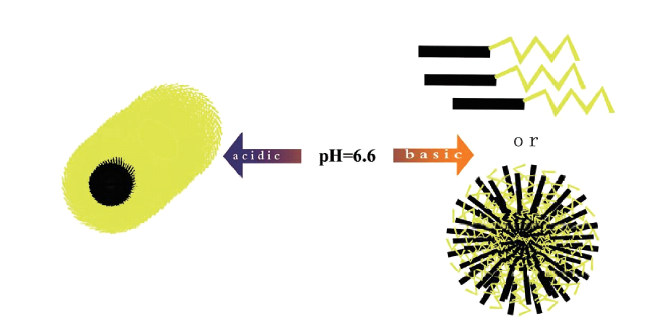
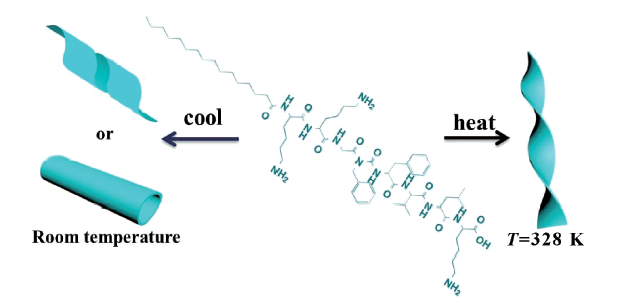
3.2 温度响应型肽自组装
温度作为刺激源,变化容易控制,而且在生物体内外的应用都比较容易操作。同时,其对形成具有β-折叠网络的纳米纤维具有重要意义[64, 65],对肽自组装的结构有很大的影响[66]。通过控制温度可以改变肽自组装驱动力(如氢键、静电作用、疏水作用等)的大小进而调控肽自组装。这个可以借助热力学的理论来解释。从热力学的观点来看,任何一个等温、等压、非体积功为零的自发过程,是由吉布斯自由能变(ΔG)决定的,ΔG又可以分解成焓变(ΔH)和熵变(ΔS)两部分:ΔG=ΔH-TΔS。任何一个上述条件下的自发过程,对应的ΔG都是负的,但却有不同的热力学特征描述:焓驱动或熵驱动。其中,焓驱动对应的是两个相互作用的分子之间特异性和反应强度的反映,包括静电作用力、氢键等。而疏水作用会使分子的疏水表面在极性环境中隐藏起来,进而使疏水分子自发地发生聚集,导致系统混乱度增加。而混乱度是系统熵大小的量度,因此,在水中两个或多个疏水分子的疏水作用是一种熵增加的自发过程,故而,疏水作用是由熵驱动的。随着温度的不断升高,分子热运动加快,系统混乱度增大,由熵驱动的疏水作用变强;同时系统焓变会增加,对于由焓驱动的相互作用,由于ΔG会增加,过程自发性减弱,导致氢键、静电相互作用的作用力减弱。
2016年,Tantakitti等[64]通过在实验室中制备PA纳米纤维,发现温度影响PA链自组装的构象,随着温度升高,形成的圆柱形纳米纤维中β-折叠结构的百分比增加。正常温度时,肽在高温下自组装的结构可以恢复原形貌。实验中将PA溶液退火至80 ℃,观察到退火所提供的能量使得具有β-折叠二级结构的纤维的成核和快速生长成为可能。随着温度升高,形成的β-折叠的总百分比也增加。Iscen等[23]在310 K、353 K和400 K时进行全原子模拟,也发现随着温度升高,形成的β-折叠的总百分比也增加。在310 K时,(20.0±0.8)%的残余物形成β-折叠,在353 K和400 K时,β-折叠的总百分比分别为(25.4±0.8)%和(27.9±1.0)%。当温度升高到400 K时,β-折叠的形成不仅出现在缬氨酸残基中,而且存在整个PA链中。
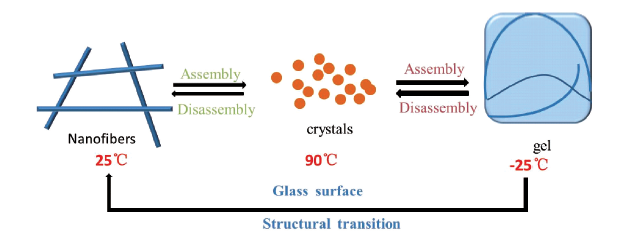
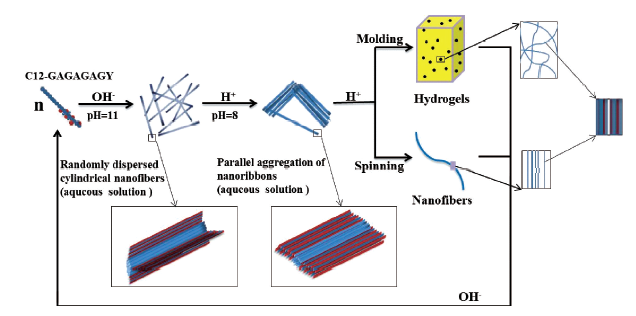
3.3 溶剂响应型肽自组装
基于多肽自组装过程的研究发现,溶剂的改变能够使多肽自组装的结构发生变化。溶剂从极性到非极性的改变使两亲性多肽分子的疏水端和亲水端在组装结构中发生反向排布,肽分子之间的疏水作用强度发生改变。而且溶剂也会诱导氨基酸残基形成氢键,促进肽自组装的形成。
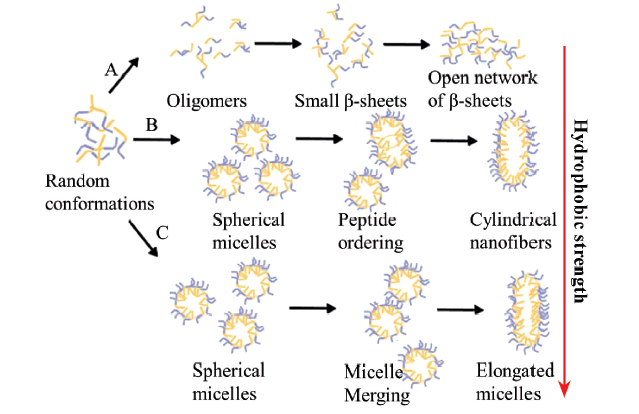
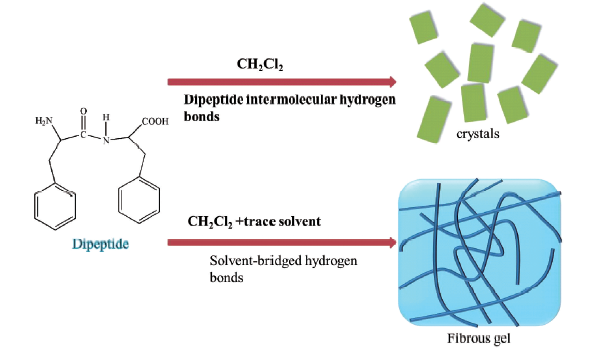
Rissanou等[71]通过模拟和实验来详细研究了FF和AA(二丙氨酸)在水和甲醇中的自组装结构。通过分布函数计算出FF和AA在甲醇和水中的含量,发现FF和AA在水中的自组装倾向明显,而在甲醇中观察到非常弱的自组装倾向。
3.4 光响应型肽自组装
光是一种清洁且可控的外部刺激,如果用于肽自组装的控制,对于设计新型肽自组装体系及相关应用具有重要的现实意义。一些分子或基团吸收了光能量后,借助所吸收的能量,使分子内或分子间产生物理性质的变化(如溶解度、颜色和导电性等)或化学反应(如聚合、二聚、异构化和光解等)。通过在肽分子主链或侧链引入感光基团,设计合成光响应型肽分子是广泛使用的方法。具有光响应性的基团的种类很多,研究最多的有:重氮或叠氮光敏基团(如邻偶氮磺酰基、偶氮苯)、光二聚型光敏基团(如香豆素、肉桂酸酯基)、丙烯酸酯基团以及特种功能的光敏基团(如具有光催化性、光致变色性和光导电性基团)等。光响应型肽分子中含有能够对外界光刺激(如近红外、紫外等)做出快速响应的光敏感基元。这些基元在适当的光波作用下会发生相应的化学、物理性质的变化,或可逆二聚(如香豆素类、肉桂酸酯类等)、或发生可逆偶极变化(如份菁类、偶氮苯类等)、或发生单向解离(如苄酯类),使自组装体微结构发生相应的转变,甚至突变,进而影响肽自组装的结构。
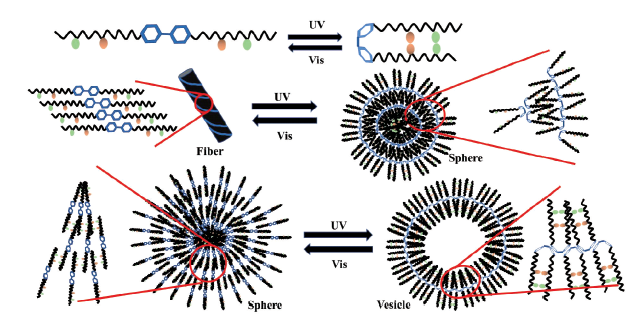
Haines等[73]利用MAX7CNB(光固化肽),在光照射的情况下,触发肽自组装成水凝胶结构。MAX7CNB在环境光下的水溶液中是展开的结构,当紫外光照射时,触发了肽的折叠,使其通过形成分子间氢键和分子内氢键形成两亲性的α-发夹构象,最终组装成水凝胶。
3.5 超声波响应型肽自组装
超声波是一种清洁能源,其主要通过破坏非共价键作用来调节肽自组装的结构形貌,在肽自组装方向上的研究还处于发展阶段。
3.6 离子响应型肽自组装
许多离子在控制多肽自组装行为方面有着重要的作用。因为离子加入后可以与个别多肽序列特异性识别并相互作用,促进肽分子的自组装,并形成特殊结构。同时,离子的加入还可以在多肽分子的极性氨基酸之间构架成盐桥,方便分子之间形成物理交联,进一步方便多肽形成稳定结构。而且,由于许多金属离子(如Cu2+、Ca2+、Zn2+等)在生物体内可以调控蛋白功能并保持体内离子的平衡等方面发挥着重要的作用。因此,离子响应型肽自组装在生物学方面有广泛的应用前景。
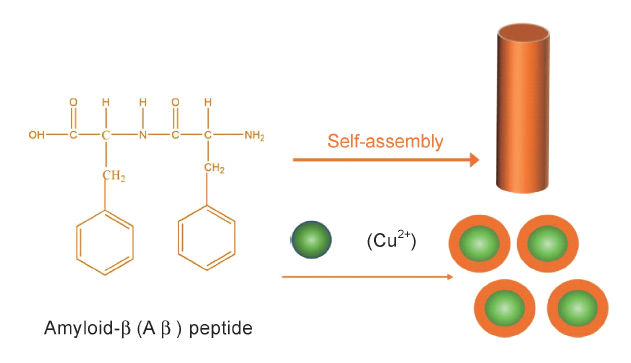
除此之外,离子强度也可以影响肽的自组装。提高环境中离子强度可以有效地屏蔽多肽分子所带的静电荷,改变多肽分子内的电荷作用,从而改变多肽的聚集形态。
Ozbas等[77]在研究多肽MAX1的自组装过程中发现,提高离子强度可以使多肽分子由无规卷曲结构自组装转变为β-折叠结构,并促使纤维网络的形成,最终可形成有一定强度的凝胶。并且,离子强度和温度越高,生成凝胶的速率和强度也会提高。
4 肽自组装的应用
4.1 药物载体和释放
2016年,Morgan等[81]通过肽自组装负载对静脉注射TF(组织因子)靶向纳米材料的方法来治疗潜在的不可压缩出血。在实验中采用大鼠经肝脏穿孔损伤的模型,通过荧光成像观察实验结果。在接受非特异性PA纳米纤维的大鼠受损肝脏中未观察到荧光。在接受结合TF因子的靶向纳米纤维的大鼠受损肝脏的血管附近观察到荧光,未受伤的部位没有观察到荧光,该现象表明靶向纳米纤维与损伤部位发生特异性结合。
4.2 组织支架促进伤口愈合
Schneider等[85]使用纳米生物技术将SAP(自组装肽)纳米纤维支架和EGF(表皮生长因子)结合来增加伤口表皮再生率。实验发现SAP经历了分子自组装能形成独特的三维结构,稳定地覆盖在伤口表面,并且EGF只在支架与人类皮肤等效性模型(HSE)直接接触时释放。通过对比试验观察伤口表面组织的闭合情况,发现与没有支架的相比,含有EGF的SAP支架伤口覆盖率加快了5倍,与没有EGF的支架相比,伤口覆盖率增加了3.5倍。
4.3 脊髓修复
2014年,Cigognini等[89]利用两种自组装肽B24和生物素-LDLK12在大鼠的脊髓挫伤中测试其对血肿和囊肿形成、生物相容性和轴突生长容许性的影响,发现可注射自组装水凝胶具有良好的止血作用并增加了再生轴突的数量,显示出了良好的生物相容性,因此自组装肽可以与药物和干细胞混合减轻血肿,提高脊髓损伤的治疗效果。
4.4 仿酶催化
与普通化学催化剂相比,天然酶是一种高效生物催化剂,然而天然酶是蛋白质,易于变性和失活,而且大部分价格昂贵。因此如何设计出易于制备、稳定、廉价、易于操作且催化活性不低于天然酶的人工酶引起了科学家极大的兴趣。
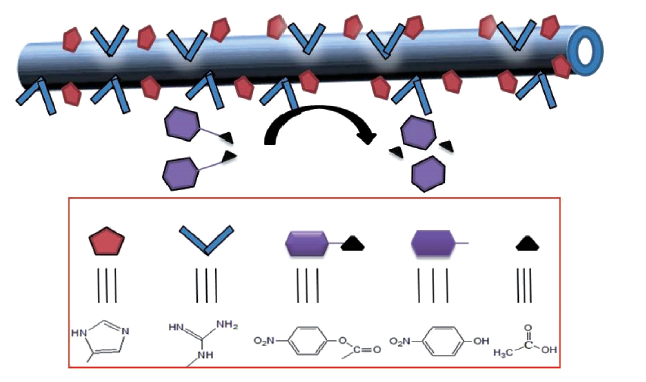
4.5 制备光学材料
研究者发现在可见光区,纳米材料并没有表现出良好的吸光度。为了克服这一缺点,他们发现在自然界中,生物分子占据了广泛的结构且在UV(紫外线)中具有天然强的CD(圆二色性)信号的构象,并且用生物分子组装成的手性纳米材料在可见光区可以人为地产生等离子基元诱导的CD信号。
Naik等[91]使用不同二级结构(无规卷曲和α-螺旋)的自组装肽人工创造光学。研究发现通过有活性的手性金纳米粒子和手性肽-纳米粒子复合物之间的相互作用,使手性肽-纳米颗粒复合物在等离子体共振频率下产生了CD信号。涂上金属离子的肽聚合体也同样产生了CD信号。光学手性是一个有吸引力的区域,可应用于各种具有可调控手性光学信号的表面等离子体纳米器件,如手性开关、超灵敏度传感器等,其在光学设备中的潜力也受到很多关注和研究。
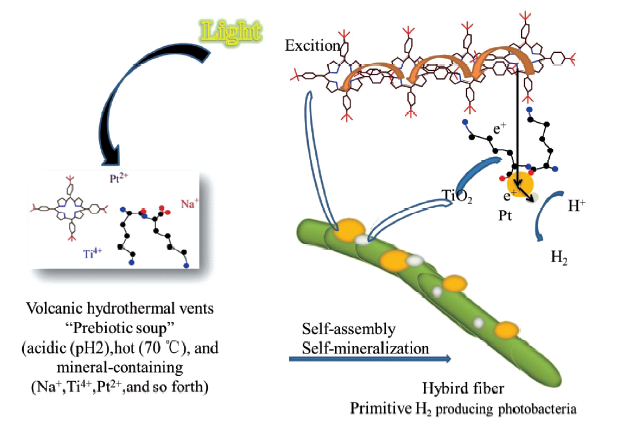
4.6 模拟光合成系统
自然光合成系统能将太阳能高效转化为化学能,模拟和创建这类系统为人们更大程度地利用太阳能提供了机遇。在自然界中的光合成系统中,最有效实现光捕获以及能量转移的方式是利用蛋白质/肽与具有发色基团的、功能性的分子自组装形成系统单元,但是其稳定性、动态属性和与特定环境的精准调控成为关键的科学问题。
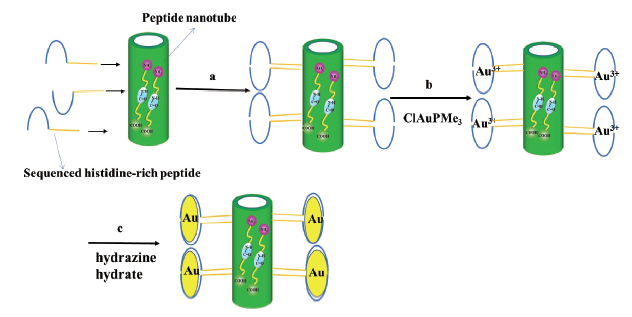
4. 生物模板
借鉴自然界的生物矿化现象,采用生物模板法合成金属或半导体材料得到了快速发展。其中,肽自组装体(如纳米管、纳米纤维)不仅能提供纳米级的微环境,还具有金属离子捕捉以及分子识别能力,并且具备大规模生产、单分散性、能通过更简单的实验方法获得、实用且价格合理等诸多优点,因此被认为是一种极具潜力的模板材料。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图15 制造金纳米线示意图(a)将含组氨酸序列的肽固定在模板的酰胺结合位点上(b)在测序组氨酸的肽上固定Au离子(c)Au纳米晶体生长[93]Fig.15 Scheme of the Au nanowire fabrication (a)Histidine sequence-containing peptides are immobilized on the amide binding site of the template (b) Au ion immobilization on the sequenced histidine-rich peptide and (c) Au nanocrystal growth[93] |
5 结论及展望
肽自组装是近几年来备受关注的热点领域,目前在基础研究和应用研究领域都取得了突破性的进展和重要成就,包括肽自组装概念的提出、肽自组装机理的研究、具有不同结构或形貌的肽自组装体的获得、肽自组装结构或形貌的可控及可调性的掌握、以及对肽自组装应用的初步探索。人类目前已经能够在一定程度上控制肽自组装,制备一些稍复杂的结构或功能的有序组装体,并且,肽自组装形成的纳米纤维、纳米管、囊泡及胶束等微纳尺度的材料,因其具有易自组装、结构稳定、生物相容性良好、可生物降解且降解产物易被吸收代谢等优点,可广泛应用于纳米药物、药物传递、骨组织损伤修复构建、仿酶催化、模拟仿光合成系统、合成模板等方面。通过肽自组装技术可以使药物达到靶向释放,还可以作为载体或者和药物、组织因子形成药物复合物来减少健康细胞受到的损害,使癌细胞或其他受伤细胞有更好的治疗效果。另外,肽通过自组装可以设计成不同的模型来帮助受损皮肤的恢复和骨组织的形成,还能够模仿生物的自然现象如模仿光合成、仿生物酶催化和合成模板等。
但是肽自组装仍然面临很多挑战:(1)自组装机制需进一步阐明,自组装结构受外界影响较大,温度、pH、溶剂、光照等都能够影响肽自组装的结构,如何精准把控外界因素得到理想的自组装结构需要科研工作者进一步研究;(2)应深入研究如何通过把控肽自组装过程中的细节提高肽自组装体在复杂环境中的稳定性,并进一步拓展肽自组装体的特殊功能;(3)和自然界中高度有序且复杂的自组装结构相比,当前对自组装的研究显得过于简单, 2005年7月初,美国Science杂志在其125周年之际,提出了“我们能够推动化学自组装走多远”的问题,指出将来对自组装的研究应进一步增加复杂程度,并多向自然界学习;因此,在肽自组装研究中,需要研究新的合成方法制备新型类肽单体,同时研究新的聚合方法构筑新的结构、新性能的高分子新材料,增加其复杂程度,更大发挥肽自组装在不同领域的应用;(4)当前肽自组装的研究与生命科学领域的交叉程度很低;自组装领域的著名学者Whitesides等认为,研究自组装的最终目的之一是了解和模拟自然界中生物的某些过程,由此可见,要使肽自组装逐步走向成熟,需要加强肽自组装的研究与生命科学领域的交叉,模仿自然界中的自组装结构,模仿自然界中精准的组装规律,模仿自然界中自主、循环、可逆的组装过程,实现自然界中生命体的某种功能。
最终期望利用肽分子组装的思想,用简便易行的方式构筑具有特殊物理和化学性质的功能组装体系,并探求和拓展其在仿生功能材料领域的应用,甚至进而超越模拟的层次,使人们在未来有可能创造出某些低级的生命形式,从而为人类认识生命现象的本质提供新方法和新思路。