





1 引言
2 以铁为始——ZVI及其改性材料
2.1 ZVI结构与污染物去除

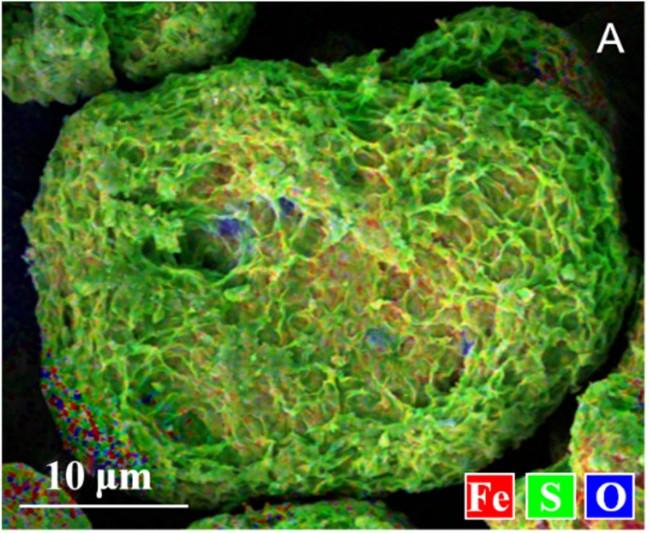
2.2 硫改性ZVI结构及改性原理
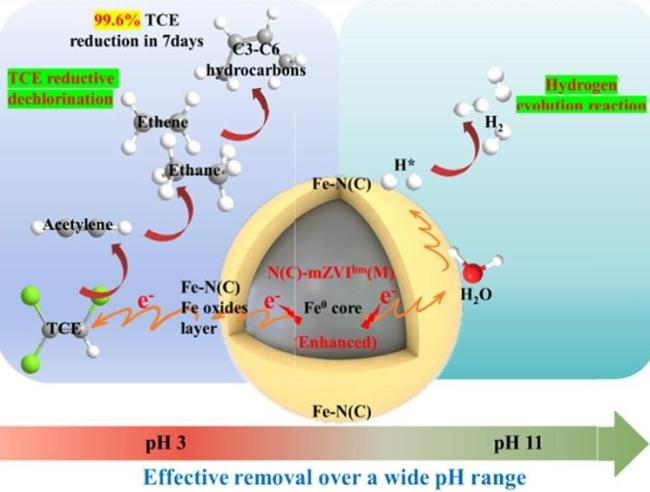
2.3 碳改性ZVI结构及改性原理
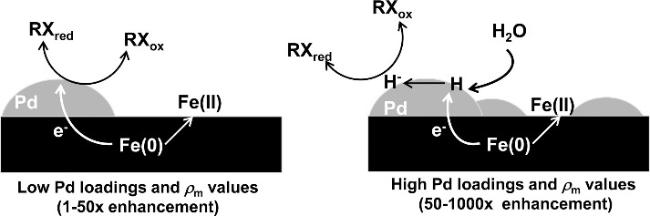
2.4 过渡金属改性ZVI结构及改性原理
3 初见端倪——还原路径及其检测手段
3.1 DET还原路径
3.1.1 电子传输
3.1.2 电子接收与还原
3.1.3 检测手段
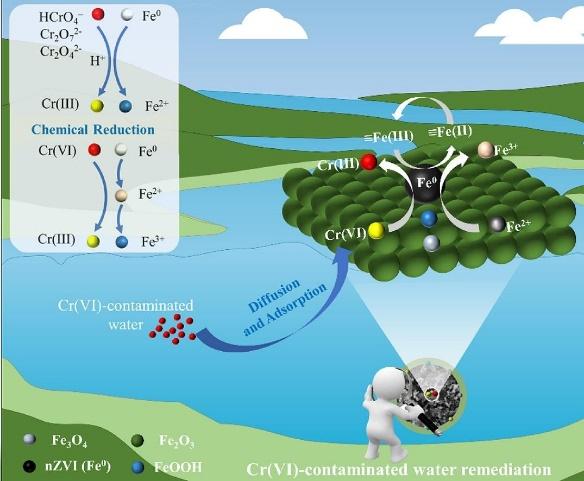
3.2 Fe(Ⅱ)还原路径
3.2.1 Fe(Ⅱ)来源
3.2.2 还原过程
3.2.3 检测手段
3.3 H*还原路径
3.3.1 析氢活性点位
3.3.2 加氢还原与产氢气
3.3.3 检测手段
4 冲突具现——表面改性对还原路径的影响
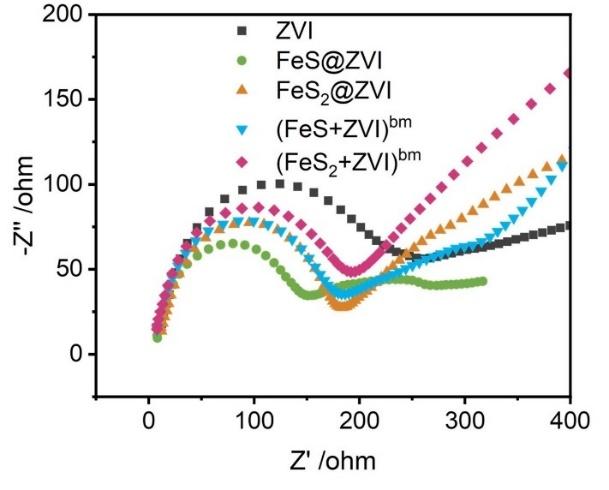
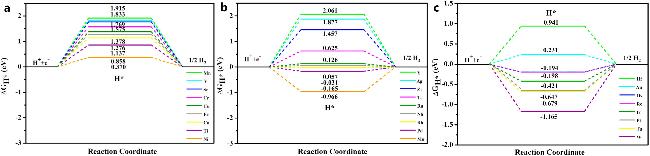
4.1 硫改性
表1 ZVI基材料的主导还原路径及其检测方法汇总Table 1 Summary of dominant reduction pathways and their detection methods for ZVI-based materials |
| Materials | Dominant Reduction Pathways | Detection Methods | Pollutants | Condition parameters | Ref |
|---|---|---|---|---|---|
| ZVI/HA | DET | TBA Quenching Experiment, Phen Experiment, KBrO3 Quenching Experiment | PNP | pH = 5, T = 25 ℃, 10 mg/L PNP, 0.5 g/L ZVI | 66 |
| LZVI | Fe(II) | Phen Experiment | Cr(VI) | pH = 6.28, T = 25 ℃, 2 mg/L Cr(VI), 5 g/L LZVI | 2 |
| ZVI | H* | Tafel, EIS, CV | CT | pH = 0.4, 30 mg/L CT | 4 |
| nZVI | H* | TBA Quenching Experiment | FF | pH = 7.0, T = 25 ℃, 0.28 mmol/L FF, 1.0 g/L nZVI | 10 |
| Fe0-Fe3O4-BM | DET and Fe(II) | Tafel, EIS, CV, Phen Experiment | Cr(VI) | pH = 3, 30 mg/L Cr(VI), 1.0 g/L Fe0-Fe3O4-BM | 53 |
| OX-ZVIbm | DET and Fe(II) | Tafel, EIS, EPR, Methanol Quenching Experiment, Phen Experiment | TCE | T = 25 ℃, 20 mg/L TCE, 4 g/L OX-ZVIbm | 12 |
| nZVI | DET and Fe(II) | Phen Experiment | Cr(VI) | pH = 6.28, T = 35 ℃, 8 mg/L Cr(VI), 0.15 g/L nZVI | 3 |
| S-nZVI | DET | Tafel | PCE | pH = 6.5, T = 25 ℃, 64 μmol/L CE, 1 g/L S-nZVI(S/Fe = 0.007) | 88 |
| S-nZVI | DET | Tafel, Kinetic Analysis | TCE | pH = 8, 10 mg/L TCE, 1 g/L S-nZVI(S/Fe = 0.20) | 6 |
| S-ZVI | DET | Solvent Kinetic Isotope Experiment | c-DCE, TCE | pH = 7.0, 0.1 mmol/L c-DCE/TCE, 5 g/L S-ZVI(S/Fe = 0.10) | 14 |
| S-mZVI | DET | Kinetic Analysis | TCE | pH = 7.0, 100 μmol/L CE, 0.26 g S-mZVI(S/Fe = 0.10) | 42 |
| S-mZVI | Fe(II) | Phen Experiment | Cr(VI) | pH = 6.00, 20 mg/L Cr(VI), 0.4 g/L S-mZVI(S/Fe = 0.112) | 105 |
| S-nZVI | H* | Tafel | VC, c-DCE | pH = 6.5, T = 25 ℃, 64 μmol/L CE, 1 g/L S-nZVI(S/Fe = 0.007) | 88 |
| S-nZVI | H* | CV, EPR, Solvent Kinetic Isotope Experiment | DTA | pH = 7.0, T = 25℃, 30 μmol/L DTA, 2.0 g/L S-nZVI(S/Fe = 0.25) | 35 |
| S-mZVI | H* | Kinetic Analysis | c-DCE, t-DCE, VC | pH = 7.0, 100 μmol/L CE, 0.26 g S-mZVI(S/Fe = 0.10) | 42 |
| S-nZVI | H* | Kinetic Analysis | TCE | pH = 7.8-8.2, T = 22 ℃, 25 mg/L TCE, 5 g/L S-nZVI(S/Fe = 0.05) | 87 |
| S-ZVI | DET and Fe(II) | Tafel, CV | c-DCE | pH = 3.0, 2 mg/L c-DCE, 0.25 g/L S-ZVI(S/Fe=0.11) | 33 |
| ZVI/FeS2 | DET and Fe(II) | Phen Experiment | NB | pH = 6.0, 25.0 mg/L NB, 0.5 g/L ZVI + 2.0 g/L FeS2 | 106 |
| S-nZVI | DET and H* | TBA Quenching Experiment | FF | pH = 7.0, T = 25 ℃, 0.28 mmol/L FF, 1.0 g/L S-nZVI(S/Fe = 0.07) | 10 |
| S-mZVI | DET and H* | TBA Quenching Experiment | CAP | T = 25 ℃, 40 mg/L CAP, 0.4 g/L S-mZVI(S/Fe = 0.112) | 11 |
| S-mZVI | DET and H* | Kinetic Analysis | PCE, 1,1-DCE | pH = 7.0, 100 μmol/L CE, 0.26 g S-mZVI(S/Fe = 0.1) | 42 |
| nZVI/CNT | H* | CV, EPR, Solvent Kinetic Isotope Experiment | Cr(VI) | pH = 6.5, T = 25 ℃, 10 mg/L Cr(VI), 0.2 g/L nZVI/CNT | 35 |
| AC-ZVI | H* | Electrochemical Analysis | TCE | pH = 8.7~9.2, T = 22 ℃, 6 mg/L TCE, 5 g/L AC-ZVI | 96 |
| AC-ZVI | H* | Electrochemical Analysis | DDT | pH = 8.8, 5 mg/L DDT, 667 g/L AC-mZVI | 15 |
| nZVI@CP-BC | DET and Fe(II) | pH Analysis, ORP Analysis | Cr(VI) | pH = 5.1, T = 25 ℃, 20 mg/L Cr(VI), 37.5 mg nZVI@CP-BC | 70 |
| BC-nZVI | DET and H* | Tafel, EIS, HER Analysis | TCE | 25 mg/L TCE, 2 g/L BC-nZVI | 29 |
| MNBC-ZVI | DET and H* | CV, TBA Quenching Experiment | TMX | T = 25 ℃, 10 mg/L TMX, 0.75 g/L MNBC-ZVI | 34 |
| nZVI/BC | DET and H* | Solvent Kinetic Isotope Experiment | TBBPA | pH = 7, T = 25 ℃, 10 mg/L TBBPA, 2.0 g/L nZVI/BC | 27 |
| nZVI@CBC | Fe(II) and H* | XPS Analysis | Cr(VI) | pH = 7.0, 4 mg/L Cr(VI), 2.5 g/L nZVI@CBC | 16 |
| Pd/Ni-nZVI | H* | Tafel, Kinetic Analysis | TCE | pH = 8, 10 mg/L TCE, 1 g/L Pd/Ni-nZVI | 6 |
| Cu-Febm(CuSO4) | H* | CV, ESR, Methanol Quenching Experiment | TCE | pH = 7, T = 25 ℃, 10 mg/L TCE, 2.0 g/L Cu-Febm(CuSO4) | 103 |
| Fe-Pd-Cu | H* | ESR | DCF | T = 25 ℃, 20 mg/L DCF, 30 g/L Fe-Pd-Cu | 51 |
| Pd/nZVI | H* | Solvent Kinetic Isotope Experiment | 1,1,1,2,5-TeCA | pH = 8, 75 μmol/L 1,1,1,2,5-TeCA, 0.22 g/L Pd/nZVI | 5 |
| Pd/nZVI | H* | Solvent Kinetic Isotope Experiment | c-DCE | pH = 8, 75 μmol/L c-DCE, 0.22 g/L Pd/nZVI | 5 |
| N(C)-mZVI | DET | Tafel, EIS, CV, TBA Quenching Experiment, | TCE | pH = 7, T = 25℃, 76 μmol/L TCE, 10 g/L mZVI | 13 |
| MoS-mZVI | DET | CV | TCE | T = 25℃, 10 mg/L TCE, 5.2 g/L MoS-ZVI | 107 |
| S-nZVI/BC | Fe(II) | Phen Experiment | Cd(II), As(III) | pH = 5.0, 20 mg/L Cd(II), 40 mg/L As(iii), 0.2 g/L S-nZVI/BC | 18 |
| Ni-nZVI/BC | H* | EIS, CV, EPR, Methanol Quenching Experiment | TCE | pH = 7.0, T = 25 ℃, 20 mg/L TCE, 1.0 g/L Ni-nZVI/BC | 17 |
| S-mZVI/GO | H* | CV | TCE | T = 25 ℃, 10 mg/L TCE, 4 g/L S-mZVI/rGO | 24 |
| Fe0@Fe-N4-C | H* | EPR | TCE | pH = 6.5, T = 25 ℃, 1 mg/L TCE, 1 g/L Fe0@Fe-N4-C | 76 |
| Ni/S-mZVI | H* | ESR | ATZ | T = 12 ℃, 4 mg/L ATZ, 24 g/L Ni/S-mZVI | 101 |
| S-mZVI/BC | DET and Fe(II) | Phen Experiment, ICP Analysis | Cr(VI) | pH = 2, 50 mg/L Cr(VI), 1 g/L S-mZVI/BC | 40 |
| NG/nZVI | DET and H* | TBA Quenching Experiment | VC | 44.8 μmol/L VC, 2 g/L nZVI, 4 g/L NG | 47 |
| S-nZVI/BC | DET and H* | TBA Quenching Experiment, Solvent Kinetic Isotope Experiment | TBBPA | pH = 7, T = 25 ℃, 10 mg/L TBBPA, 2.0 g/L S-nZVI/BC(S/Fe = 0.090) | 108 |
| S-nZVI/BC | DET and H* | Solvent Kinetic Isotope Experiment | TBBPA | pH = 7, T = 25 ℃, 10 mg/L TBBPA, 2.0 g/L S-nZVI/BC | 27 |
| S-nZVI@NBC | DET and H* | DFT Analysis, PDOS Analysis | NOR | 10 mg/L NOR, 0.8 g/L S-nZVI@NBC | 81 |
| BC@Fe/Ni | DET and H* | XPS Analysis | 2,4-DCP | T = 25 ℃, 50 mg/L 2, 4-DCP, 2 g/L BC@Fe/Ni | 109 |
*Full form of partial abbreviations: activated carbon (AC), carbon nanotube (CNT), biochar (BC), tertiary-butyl alcohol (TBA), oxidation-reduction potential (ORP), inductively coupled plasma (ICP), density functional theory (DFT), partial density of states (PDOS), p-nitrophenol (PNP), carbon tetrachloride (CT), florfenicol (FF), trichloroethylene (TCE), perchlorethylene (PCE), dichloroethylene (DCE), vinyl chloride (VC), diatrizoic acid (DTA), nitrobenzene (NB), chloramphenicol (CAP), dichlorodiphenyltrichloroethane (DDT), thiamethoxam (TMX), tetrabromobisphenol A (TBBPA), tetrachloroethane (TeCA), atrazine (ATZ), norfloxacin (NOR), dichlorophenols (DCP) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}