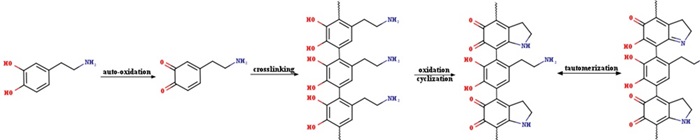
1 引言
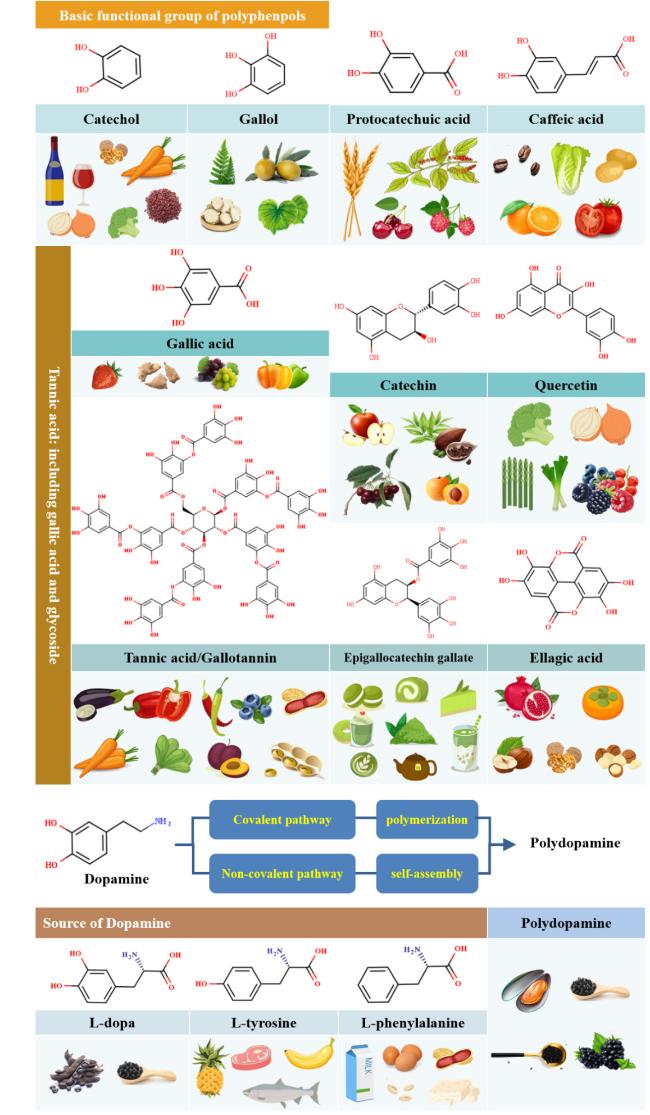
表1 多酚类化合物的分类与基本理化性质Table 1 Classification and basic physicochemical properties of polyphenols |
| Classification | Name | CASa) | Molecular formula | MWb) (g/mol) | Log KOWc) | Log KAWc) | Log KOAc) | WS (mg/mL)d) | pKa (pH=0~14)e) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phenolic acids and derivatives | ||||||||||||
| Hydroxycinnamic acids | Chlorogenic acid | 327-97-9 | C16H18O9 | 354.32 | -1.01 | -20.29 | 19.28 | 404.70 | 3.33 | 9.21 | 12.47 | - |
| Caffeic acid | 331-39-5 | C9H8O4 | 180.16 | 1.11 | -14.24 | 15.39 | 52.07 | 3.84 | 9.28 | 12.69 | - | |
| Ferulic acid | 1135-24-6 | C10H10O4 | 194.19 | 1.42 | -11.49 | 13.00 | 5.97 | 3.97 | 9.98 | - | - | |
| Hydroxybenzoic acids | Gallic acid | 149-91-7 | C7H6O5 | 170.12 | 0.86 | -17.3 | 18.00 | 58.95 | 3.94 | 9.04 | 11.17 | - |
| Protocatechuic acid | 99-50-3 | C7H6O4 | 154.12 | 0.91 | -13.32 | 14.18 | 50.98 | 4.16 | 9.40 | 12.84 | - | |
| Derivatives | Protocatechualdehyde | 139-85-5 | C7H6O3 | 138.12 | 0.75 | -11.22 | 12.32 | 43.39 | 7.84 | 11.90 | - | - |
| 3,4-Dihydroxyacetophenone | 1197-09-7 | C8H8O3 | 152.15 | 0.71 | -11.36 | 12.07 | 17.44 | 7.90 | 11.88 | - | - | |
| Hydroxytyrosol | 10597-60-1 | C8H10O3 | 154.17 | 0.61 | -12.89 | 13.5 | 271.60 | 9.55 | 12.99 | > 14 | ||
| 3,4-Dihydroxyphenylglycol | 3343-19-9 | C8H10O4 | 170.17 | -0.53 | -14.33 | 13.32 | 1000.00 | 9.21 | 12.63 | 13.92 | > 14 | |
| Tannic acid/Gallotannin | 1401-55-4 | C27O24H18 | 636.48 | -0.19 | - | -999.00 | 0.51 | ≥ 7.41 | ||||
| Ellagic acid | 476-66-4 | C14H6O8 | 302.20 | -0.52 | - | -999.00 | 33.35 | 5.54 | 6.22 | 11.78 | 12.40 | |
| Polyphenolic Amides | ||||||||||||
| Catecholamines | Dopamine | 51-61-6 | C8H11NO2 | 153.18 | 0.38 | -12.44 | 11.46 | 1000.00 | 9.31 | 9.99 | 13.03 | - |
| Levodopa/L-dopa | 59-92-7 | C9H11NO4 | 197.19 | -2.24 | -16.28 | 13.53 | 320.10 | 1.65 | 9.06 | 9.69 | 12.74 | |
| Flavonoids | ||||||||||||
| Flavones | Chrysin | 480-40-0 | C15H10O4 | 254.24 | 3.32 | -10.7 | 14.22 | 84.00 | 5.43 | 7.03 | - | - |
| Flavonols | Quercetin | 117-39-5 | C15H10O7 | 302.24 | 1.48 | -18.57 | 20.05 | 2.47 | 5.22 | 6.69 | 7.78 | 9.46&12.82 |
| Myricetin | 529-44-2 | C15H10O8 | 318.24 | 1.42 | -22.55 | 23.97 | 2.23 | 5.52 | 6.64 | 7.64 | 9.13&11.33 | |
| Rutin | 153-18-4 | C27H30O16 | 610.53 | -2.02 | -36.89 | 34.87 | 27.56 | 5.23 | 6.85 | 8.56 | 11.73-13.53 | |
| Flavanonols /Dihydroflavonols | Taxifolin/Dihydroquercetin | 480-18-2 | C15H12O7 | 304.25 | 0.59 | -19.71 | 20.66 | 22.03 | 7.74 | 9.00 | 9.61 | 12.16 |
| Flavanones | Hesperitin | 520-33-2 | C16H14O6 | 302.29 | 2.44 | -16.15 | 18.75 | 0.27 | 7.86 | 9.33 | 9.98 | - |
| Flavanols | Procyanidin | 4852-22-6 | C30H26O13 | 594.52 | 2* | - | - | 0.39** | 8.65 | 9.09 | 9.45&9.86 | ≥ 10.59 |
| Catechin/ Cianidanol | 154-23-4 | C15H14O6 | 290.27 | 1.18 | -23.54 | 24.05 | 63.11 | 9.04 | 9.69 | 10.89 | 12.65 | |
| Chalcones | Butein | 487-52-5 | C15H12O5 | 272.26 | 2.51 | -17.85 | 20.36 | 0.48 | 7.75 | 8.77 | 9.35 | 12.46 |
| Dihydrochalcones | Phloretin | 60-82-2 | C15H14O5 | 274.28 | 3.51 | -14.1 | 17.61 | 0.07 | 8.00 | 9.49 | 10.68 | 11.96 |
| Anthocyanins | Delphinidin | 528-53-0 | C15H11O7 | 338.70 | 2.14 | - | -999.00 | 0.41 | - | |||
| Cyanidin | 528-58-5 | C15H11O6 | 322.70 | 2.2 | - | -999.00 | 0.45 | - | ||||
| Others | ||||||||||||
| Coumarins | 6,7-Dihydroxycoumarin/ Esculetin | 305-01-1 | C9H6O4 | 178.15 | 0.55 | -11.51 | 12.06 | 72.13 | 7.91 | |||
| Daphnetin | 486-35-1 | C9H6O4 | 178.15 | 0.55 | -11.51 | 12.06 | 72.13 | 8.05 | ||||
Annotation(1) AbbreviationaCAS = Chemical Abstracts Service Number,which are all acquired from Pub Chem (https://pubchem.ncbi.nlm.nih.gov/).bMW = Molecular Weight,which are acquired from Pub Chem computed by PubChem 2.2 and Chemical Book (https://www.chemicalbook.com).cKow = Octanol-water partitioning coefficient,cKOA = Octanol-air partitioning coefficient, cKAW = Air-water partitioning coefficient,which are all generated using the US Environmental Protection Agency’s EPI Suite. Besides,Kow is also computed by XLogP3-AA using XLogP3 3.0 (*). dWS = Water Solubility at 25 ℃,which are generated using the US Environmental Protection Agency’s EPI Suite and the Chemaxon’s ChemAxon Marvin Suite (**).epKa = Dissociation Constant at 25 ℃,which are all generated using the Chemaxon’s ChemAxon Marvin Suite.(2) Classification: derived from Refs. [20,22 -25] and Phenol-Explorer 3.6 (http://phenol-explorer.eu/).(3) Name: Referred to Pub Chem,Chemical Book and Phenol-Explorer 3.6. |
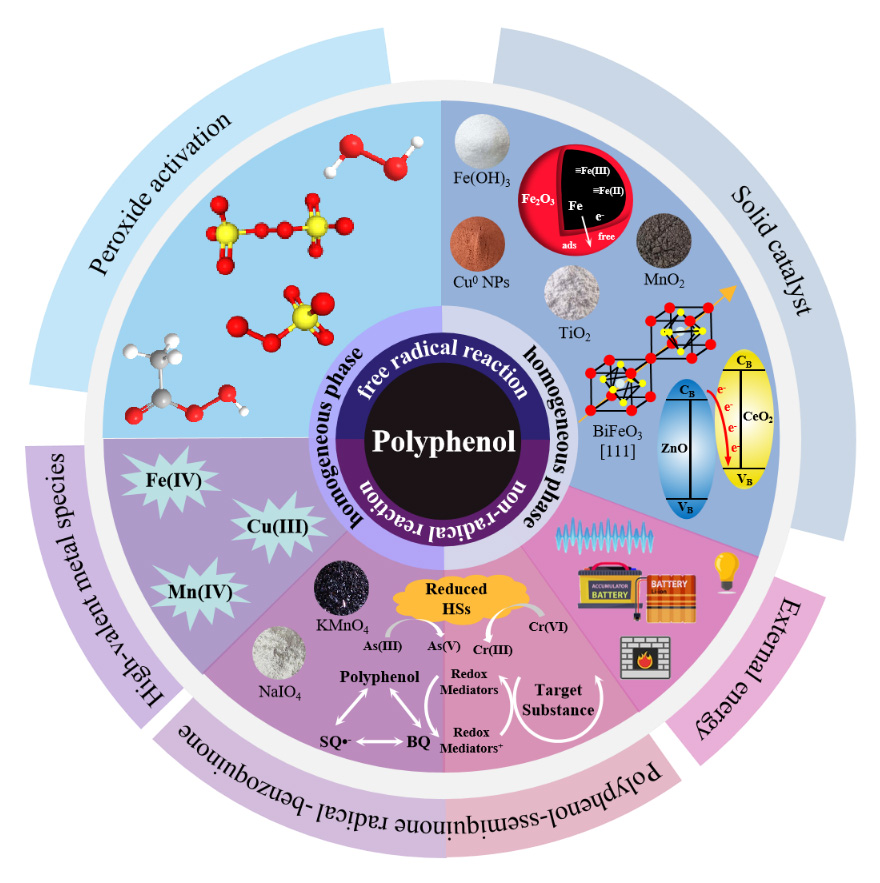
2 过氧化氢/过硫酸盐/过氧乙酸的活化
2.1 诱导过氧化氢/过硫酸盐/过氧乙酸产生活性氧物种
2.2 形成多酚/铁或铜离子/过氧化物体系
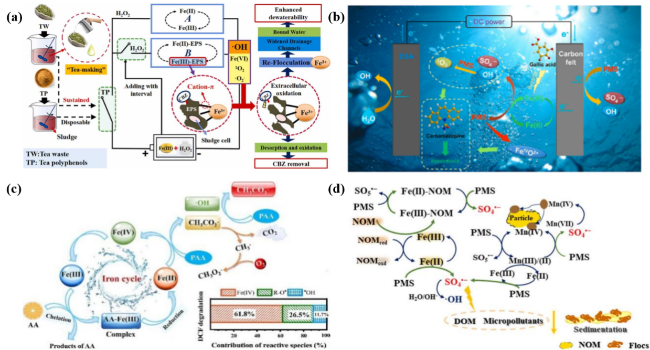
图2 多酚类化合物在各类典型多酚/铁或铜离子/过氧化物体系中的反应机制:(a) 调控H2O2投加间隔催化TPs/Fe(III)/H2O2持续产生ROS去除卡马西平(Carbamazepine,CBZ)和EPS[97];(b) GA参与电催化PMS去除CBZ等[102];(c) AA参与催化活化PAA过程所生成的Fe(IV)主导双氯芬酸(Diclofenac,DCF)的去除[144];(d) NOM-多金属参与的PMS多种活化反应路径[141]Fig. 2 Reaction mechanisms of polyphenolic compounds in typical polyphenols/Fe(Cu) ion/peroxide systems. (a) Controlled H2O2 dosing catalyzes TPs/Fe(III)/H2O2 to continuously produce ROS and remove CBZ and EPS[97];(b) removal of CBZ by electrocatalytic activation of PMS with GA[102];(c) AA participated in the catalytic activation of PAA to produce Fe(IV) dominated the removal of DCF[144];(d) multiple activation reaction pathways of PMS involving NOM-multimetals[141] |
表2 天然多酚参与的芬顿/类芬顿反应Table 2 Natural polyphenols involved Fenton/Fenton-like reactions |
| Natural polyphenols | Catalyst | Oxidizing agent | External energy or substances | Active species | Water pollutants | Research objectives | Ref | |
|---|---|---|---|---|---|---|---|---|
| Pollutant type | Pollutant name | |||||||
| Catechin | Fe(ClO4)3·H2O | H2O2 | - | •OH,O2•- | Pharmaceuticals and personal care products | Atenolol | Degradation | 124 |
| Tannin extract of black wattle | FeSO4·7H2O | O2,H2O2 | •OH,HO2•/O2•- | Amino acids | Methionine | Degradation | 109 | |
| Catechin | Fe(ClO4)3·xH2O | H2O2 | - | •OH, O2•-,SQ•- | Endocrine disrupters | Bisphenol A | Degradation | 93 |
| Phenol,Catechol,Resorcinol and Hydroquinone | FeCl3·6H2O | H2O2 | - | •OH,O2•- | Phthalate esters | Dimethyl phthalate | Degradation | 125 |
| Catechin | FeCl3 | H2O2 (self-generation),O2 | simulated sunlight | •OH,HO2•/O2•-,H2O2 | Pharmaceuticals and personal care products | Inderal | Degradation | 96 |
| Tea leaves and coffee grounds (Caffeic acid,L (+)-Ascorbic acid,Gallic acid,Catechin,Chlorogenic acid) | FeCl3, FeCl2·4H2O | H2O2 | near-UV light irradiation | •OH | Dyestuffs | Methylene blue | Degradation | 94 |
| Gallic acid | FeSO4·7H2O, Fe2(SO4)3·xH2O | H2O2,O2 | - | •OH,O2•- | Pesticides | Pentachlorophenol | Degradation | 126 |
| Gallic acid,Catechol, 3,4-Dihydroxyphenylaceticacid, 2,3-Dihydroxybenzoic acid, 2,5-Dihydroxybenzoic acid | Fe(NO3)3,FeSO4 | H2O2 | - | •OH | Dyestuffs | Bismarck brown Y | Degradation | 127 |
| Gallic acid | FeCl3 | H2O2 | - | •OH | Dyestuffs | Methyl orange | Discoloration | 128 |
| Tannic acid | FeSO4·7H2O, Fe2(SO4)3·9H2O | H2O2 | - | •OH,HO2•/O2•-,SQ•- | Pesticides | 2,4,6-Trichlorophenol | Degradation | 129 |
| Protocatechuic acid | FeSO4·7H2O, Fe(NO3)3·9H2O | H2O2 | - | •OH | Pesticides | Alachlor | Degradation | 95 |
| Olive mill wastewater (including polyphenols) | Fe2(SO4)3·H2O | H2O2 | solar irradiation | •OH,HO2•/O2•- | Pesticides | Terbutryn,Diclofenac,Chlorfenvinphos,Pentachlorophenol | Discoloration | 130 |
| Olive mill wastewater (including polyphenols) | FeSO4·7H2O, Fe2(SO4)3·H2O | H2O2 | irradiation by a Xe lamp | •OH | Food additives (artificial sweetener) | Saccharin | Degradation | 131 |
| Cork boiling wastewater (Gallic acid,Tannic acid and Phenol) | Fe2(SO4)3·xH2O | H2O2 | solar simulator under constant illumination from a Xenon lamp | •OH | Pesticides | Imidacloprid,Methomyl | Degradation | 132 |
| The extracts of Theobroma grandiflorum (total polyphenols,Citric acid,Ascorbic acid) | FeSO4·7H2O | H2O2 | sunlight | •OH | Pharmaceuticals and personal care products | Acetaminophen, Diclofenac, Ciprofloxacin,Sulfamethoxazole | Photocatalytic, Electrocatalytic degradation | 97 |
| electrode and electrolyte | Cl• | |||||||
| Ascorbic acid (non-polyphenol) | FeSO4·7H2O, Fe2(SO4)3 | CaO2 (H2O2, self-generation), O2 (self-generation) | - | •OH,HO2•/O2•- | Chemical materials | Trichloroethene | Degradation | 99 |
| Ascorbic acid (non-polyphenol) | Cu(NO3)2·9H2O | H2O2 (self-generation),O2 | - | •OH,Cu(III), HO2•/O2•-,H2O2 | Pharmaceuticals and personal care products | Sulfamethoxazole | Degradation | 133 |
| - | Bacteria | Escherichia coli | Inactivation | |||||
表3 天然多酚参与的过硫酸盐反应Table 3 Natural polyphenols involved advanced oxidation processes of persulfate |
| Natural polyphenols | Catalyst | Oxidizing agent | External energy or substances | Active species | Water pollutants | Research objectives | Ref | |
|---|---|---|---|---|---|---|---|---|
| Pollutant type | Pollutant name | |||||||
| Catechin | Fe(ClO4)3·H2O | PDS (K2S2O8) | - | SO4•-,•OH | Pharmaceuticals and personal care products | Atenolol | Degradation | 124 |
| Caffeic acid | Fe2(SO4)3 | PDS(Na2S2O3) | - | SO4•-,•OH , SO5•-、Fe(IV) | Endocrine disrupters | Bisphenol A | Degradation | 114 |
| Gallic acid | Fe(ClO4)3·xH2O, Fe(NH4)2·(SO4)2·6H2O | PDS (Na2S2O8),O2 | - | SO4•-,•OH, HO2•/O2•- | Pharmaceuticals and personal care products | Ibuprofen | Degradation | 134 |
| p-Benzoquinone | Fe(NO3)3 | PMS (KHSO5·0.5KHSO4· 0.5K2SO4) | - | SO4•-,•OH, Fe(IV) | Pesticides | Atrazine,Atrazine-desethyl, Atrazine-desisopropyl | Degradation | 135 |
| Catechin | FeCl3O12·xH2O | PMS (2KHSO5·KHSO4·K2SO4) | - | SO4•-,Fe(IV), 1O2, | Pharmaceuticals and personal care products | Ofloxacin | Degradation | 136 |
| Catechin | Fe(ClO4)3 | PDS (Na2S2O8),O2 | - | SO4•-, •OH, HO2•/O2•-,SQ•- | Pharmaceuticals and personal care products | Naproxen | Degradation | 137 |
| Methyl-p-benzoquinone, Methyl-hydroquinone | Fe(NO3)3·9H2O, FeSO4·7H2O | PDS (Na2S2O8) | - | SO4•-,•OH, Fe(IV) | Pesticides | Atrazine,Atrazine-desethyl, Atrazine-desisopropyl | Degradation | 138 |
| Gallic acid | FeCl3·6H2O | PMS (KHSO5·0.5KHSO4· 0.5K2SO4) | - | SO4•-,•OH, HO2•/O2•- | Flame retardants | Polybrominated diphenyl ethers (BDE47) | Degradation | 104 |
| Protocatechuic acid | FeSO4·7H2O | PMS (2KHSO5·KHSO4·K2SO4) | - | SO4•-,•OH, HO2•/O2•-,1O2 | Pharmaceuticals and personal care products | Ciprofloxacin | Degradation | 74 |
| Catechin | Fe(ClO4)3 | PDS (Na2S2O8) | (UVA) | SO4•-,•OH | Pharmaceuticals and personal care products | Atenolol | Degradation | 139 |
| Epigallocatechin gallate | FeSO4·7H2O | PS (Na2S2O8, Na2S2O3·5H2O),O2 | - | SO4•-,•OH,SQ•-, | Pesticides | Atrazine | Degradation | 113 |
| Gallic acid | Fe2(SO4)3 | PMS (2KHSO5· KHSO4·K2SO4) | electrode and electrolyte | • OH,SO4•-, 1O2,Fe(IV) | Pharmaceuticals and personal care products | Carbamazepine, Sulfamethoxazole, Sulfisoxazole,Metronidazole | Degradation | 102 |
| Tannic acid | FeSO4·7H2O | PDS (Na2S2O8) | SO4•-,•OH, HO2•/O2•- | EPA priority pollutant | Trichloroethylene | Removal | 140 | |
| NOM (HAs,ascorbic acid) | KMnO₄,FeCl3 | PMS (KHSO5·0.5KHSO4· 0.5K2SO4),Na2S2O3 | • OH,SO4•-, Mn(II/III,IV, VII),NOM* | Pharmaceuticals and personal care products | Sulfamethoxazole | Coagulation,Oxidation | 141 | |
| Epigallocatechin gallate | CuCl,CuSO4·5H2O | PMS (Na2S2O3·5H2O), H2O2 (self-generation) | - | SO4•-, •OH, HO2•/O2•-,Cu(III) | Endocrine disrupters | Bisphenol A | Degradation | 142 |
| Gallic acid | CuCl,CuSO4·7H2O | PMS (KHSO5·0.5KHSO4· 0.5K2SO4),Na2S2O3 | - | SO4•-, Cu(III), •OH,1O2 | Flame retardants | Tetrabromobisphenol A | Degradation | 143 |
2.3 多酚-金属离子的螯合与还原作用

2.4 非自由基反应
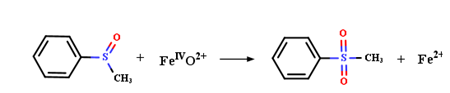
3 高价金属
3.1 铁离子

图4 高价金属催化水中典型污染物降解机理;(a) CFA和(b) MBQ参与PS氧化Fe(II)产生Fe(IV)去除BPA和ATZ及其转化中间体的详细过程[114,138];(c) 不同价态铜离子的相互转化以及Cu(III)降解TBBPA的反应机制[143];(d) 联合使用高价铁和锰提高氧化电位增强污染物去除速率[154];(e)BQ对含锰物质转化的影响及不同价态锰去除BPA的反应汇总[152]Fig. 4 Degradation mechanisms of typical pollutants in water catalyzed by high-valent metal species. Detailed process of (a) CFA and (b) MBQ involved in the oxidation of Fe(II) by PS to produce Fe(IV) for the removal of BPA and ATZ and their intermediates[114,138];(c) interconversion of Cu ions in different valence states and the reaction mechanism for the degradation of TBBPA by Cu(III)[143];(d) combined use of high-valent Fe and Mn to increase oxidation potential and enhance pollutant removal rate[154];(e) summary of the effect of BQ on the transformation of Mn-containing substances and the reactions of different valence states of Mn for the removal of BPA[152] |
3.2 铜离子
3.3 锰离子
4 固体催化剂
4.1 零价金属单体
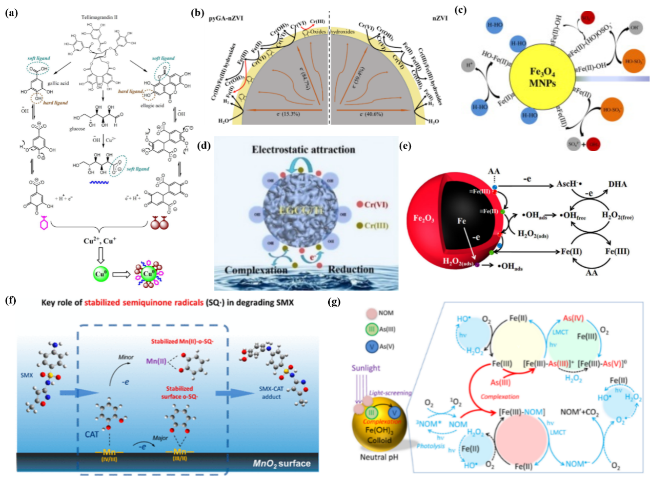
图5 (a) Hibiscus rosa-sinensis提取物制备Cu0 NPs[155];(b) pyGA-nZVI和nZVI还原Cr(VI)机制[158];(c) Fe3O4 MNPs表面可能发生的非均相反应[168];(d) EGCG修饰Ti去除Cr(VI)的螯合与还原作用[161];(e) AA/Fe3O4/H2O2非均相体系中均相芬顿反应和非均相芬顿反应对甲草胺整体降解的单独贡献[167];(f) 儿茶酚为代表的HAs在MnO2的稳定与催化作用下降解SMX的反应机制[181];(g) NOM遮光效应和竞争络合效应对CFH去除As(III)的影响[185]Fig. 5 (a) Preparation of Cu0 NPs from Hibiscus rosa-sinensis extracts[155];(b) mechanism of Cr(VI) reduction by pyGA-nZVI and nZVI[158];(c) possible heterogeneous reactions on the surface of Fe3O4 MNPs[168];(d) homogeneous and heterogeneous Fenton reactions in the heterogeneous system of AA/Fe3O4/H2O2 on the alachlor overall degradation individually[161];(e) chelation and reduction of Cr(VI) removal by EGCG-modified Ti[167];(f) the reaction mechanism of the degradation of SMX by HAs represented by catechols in the context of stabilization and catalytic effect of MnO2[181];(g) the mechanism of NOM light-screening effect and competitive complexation effect on the CFH removal of As(III)[185] |
4.2 单金属化合物
4.3 多金属化合物
+

图6 (a) NiFe2O4 NPs在光辐射下降解MB的反应机理[187];(b) CeO2@ZnO Z型异质结构建及光生载流子迁移机制[191];(c) 光辐射下TiO2@EA、(R-Vo)TiO2@EA和Nd-Fe-(D-Vo)TiO2@EA的电位以及CB、VB、LUMO和HOMO等参数[193];(d) 碳基材料(HAs-Fe@BC)联合PDS降解RhB的反应机制[195];(e) 天然多酚橡木胆单宁改性HMS的具体修饰过程[201];(f) 多酚六种常见相互作用[203];CFA在金红石(110)表面(g)和ZnO (1010)表面(h)形成桥联双齿和螯合双齿结构[204]Fig. 6 (a) Reaction mechanism of NiFe2O4 NPs for the degradation of MB under optical radiation[187];(b) Z-type heterostructure construction and photogenerated carrier migration mechanism in CeO2@ZnO[191];(c) potentials as as well as VB,CB,LUMO and HOMO parameters for the photovoltaic TiO2@EA,(R-Vo) TiO2@EA and Nd-Fe-(D-Vo)TiO2@EA potentials as well as the summary of values of valence band,conduction band,LUMO and HOMO parameters under light radiation[193];(d) reaction mechanism of carbon-based material (HAs-Fe@BC) in combination with PDS for the degradation of RhB[195];(e) specific modification process of HMS modified by natural polyphenol oak gall tannin[201];(f) six common interactions of polyphenols[203];CA on the rutile (110) surface (g) and ZnO (1010) surface (h) formation of bridging bidentate and chelating bidentate structures[204] |
4.4 金属有机配合物
4.5 碳基材料
4.6 无机盐负载型催化剂
5 多酚-半醌自由基-醌类物质
5.1 高碘酸盐和高锰酸盐
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
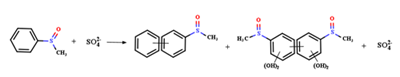
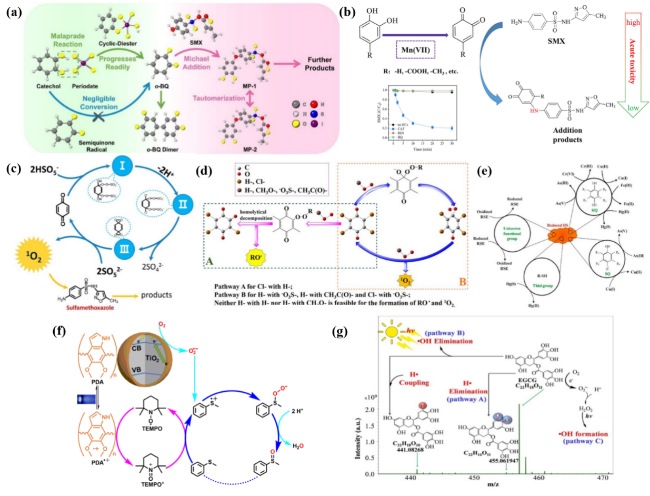
图7 多酚类物质诱导高碘酸盐(a)和高锰酸盐(b)生成活性物种选择性去除SMX、NPX、ATZ和BPA等污染物的反应机制[211-212];(c) BQ/PMS体系双环氧乙烷的生成、分解与催化生成1O2的关键步骤[215];(d) 量子化学计算描述 p-BQ、TCBQ活化典型过氧化物机制 [213];(e) HSs存在下常见RES的氧化还原反应[246];(f) LED蓝光光照下PDA与TiO2、TEMPO、O2选择性氧化硫化物的反应机制[174];(g) 太阳光照下EGCG体系中•OH和PFRs的来源与生成机制[251]Fig. 7 Reaction mechanisms of polyphenol-induced periodate (a) and permanganate (b) generation of reactive species for selective removal of pollutants such as SMX,NPX,ATZ,and BPA[211-212];(c) key steps in the generation and decomposition of diethylene oxide with catalytic generation of 1O2 in the BQ/PMS system[215];(d) quantum chemical calculations describing the mechanisms of activation of typical peroxides by p-BQ and TCBQ[213];(e) redox reactions of common RES in the presence of HSs[246];(f) reaction mechanism of selective oxidation of sulfides by PDA with TiO2,TEMPO,and O2 under LED blue light illumination[174];(g) source and generation mechanism of •OH and PFRs in EGCG system under solar illumination[251] |
5.2 过氧化物
5.3 氧气、水和其他物质
5.4 氧化还原介体