



1 引言
光子与分子作用,除了可以将分子激发到激发态,还可能使分子发生光解离或光电离。对分子的光解离和光电离过程进行深入研究,在分子反应动力学领域非常重要。
分子光解离是化学反应中的关键反应类型之一。在分子的光解离过程中,母分子首先吸收一个或多个光子跃迁到激发态,然后再解离成两个或者多个碎片。这些碎片大多是自由基或者原子,自由基碎片具有一定的内能,呈现较高的反应活性,是很多光化学反应的起始反应物。分子光解离也被称为半碰撞反应。双分子(原子)基元反应是两个分子先相互碰撞、暂时结合在一起形成一个过渡态结构,然后再分解成两个新的分子,而分子光解离过程与双分子反应中碰撞、结合、分解过程的后半部分相似。因此,研究分子光解离动力学,不仅有助于理解分子的电子激发态特性和分子被激发后的动力学过程,而且对于深入认识、理解双分子反应的机理有着重要意义[1]。通过研究多通道光解离反应动力学,可以获得产物的分支比与反应条件尤其是解离光子能量之间的联系,进而为实现对化学反应的调控提供重要的理论依据。研究分子光解离动力学在工农业生产和大气环境领域也具有重要意义。例如,氟利昂是人们生产生活中一类非常重要的制冷剂分子,它们光解离产生的卤原子会导致大气平流层产生臭氧空洞,海洋中释放的卤代烃的光解离也会加剧对臭氧层的破坏,卤代物的光解离截面会影响臭氧层破坏的光化学参数和理论模型[2⇓-4];研究卤代烃分子的光解离对治理大气污染、保护臭氧层有着重要的指导作用。分子光解离是大气化学中的关键反应,太阳的紫外辐射能使很多种大气分子发生光解离,促成大气中的多种化学反应。分子光解离还在太阳能利用等方面起着重要作用。
分子光解碎片平动能谱(Photofragment Translational Spectroscopy)是研究分子光解离反应动力学非常重要的方法[5]。该方法可以精确地测量光解离碎片的平动能分布和空间角分布,获得光解离反应的各种解离通道和通道分支比、各通道光解离碎片的量子态布居和可资用能分配,进一步分析可以获得分子激发态势能面的量子态干涉、共振、锥形交叉等非绝热相互作用对分子光解离过程的影响等,对深入理解分子光解离物理化学机制有重要的作用。朱起鹤院士团队是我国最早开展分子光解离动力学研究的课题组,他们自主研制了多台光解碎片平动能谱仪,并利用这些仪器开展了一系列小分子的光解离动力学研究,取得了具有重要科学意义的研究成果。
分子电子激发态在构型、能量、极性等方面均展现出与分子基态不同的特性,这些特性赋予它们更高的化学反应活性。利用紫外-可见吸收光谱、共振增强多光子电离(REMPI)光谱[8-9]和激光诱导荧光光谱(LIF)[10]等方法,可以获得分子电子激发态信息。REMPI光谱是目前研究激发态光谱的常用方法之一,其原理是分子吸收适配的激光光子能量共振跃迁到特定激发态后再吸收一个或者多个光子发生电离,通过测量产生的离子信号,可以获得电子激发态高分辨、高信噪比光谱。在火焰燃烧、星际介质演化、生命活动及等离子体反应等过程中,高化学活性的离子态物种起到了至关重要的作用。对离子态光谱的深入研究,能够获得其空间构型、能级结构等关键信息,能帮助我们深入理解离子在化学反应网络中所起到的重要作用。研究分子离子状态的光谱技术有零动能电子谱(ZEKE)[11]、光电子能谱(PES)[12]、质量解析阈值电离谱(MATI)[13]等。朱起鹤院士团队自主搭建了REMPI、MATI光谱仪,并采用多种红外-紫外激光结合的光谱技术,开展了一系列苯衍生物分子及其团簇的光电离微观机理研究,取得了重要进展。
本文将系统介绍朱起鹤院士团队在一些重要小分子光解离和光电离微观机理研究方面取得的成果。主要内容包括:
● 重要小分子的光解离动力学研究
光解碎片平动能谱仪的研制
卤代甲烷的光解离研究
卤代乙烷的光解离研究
其他卤代烃的光解离研究
● 重要小分子的光电离动力学研究
REMPI/MATI和IR-UV光谱实验装置
苯衍生物的REMPI和MATI光谱研究
苯衍生物分子的构象异构研究
苯衍生物及其团簇的IR/UV激光光谱研究
在此基础上,本文对相关的研究工作进行了总结和展望。
2 重要小分子的光解离动力学研究
2.1 光解碎片平动能谱仪的研制

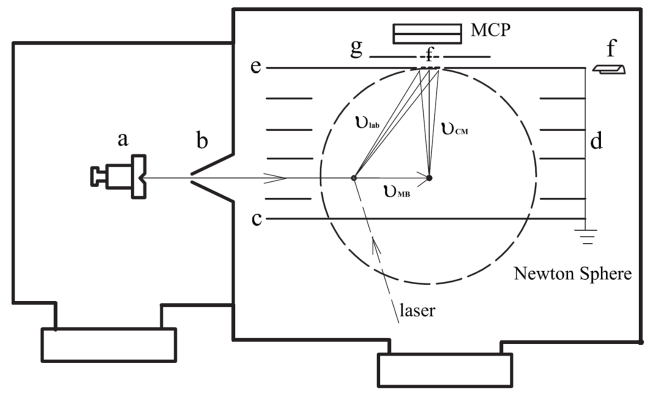
2016年,他们又采用弱电场加速短距离飞行的方案,研制成功低电压加速离子速度成像式小型光解碎片平动能谱仪[17](图3)。其核心技术在于巧妙地将低电压加速、短距离飞行的设计思路与传统的离子透镜聚焦方法相结合。采用了较低的电压(30~150 V)代替传统的高电压(650~4000 V)加速和聚焦光解碎片离子,离子飞行的距离仅有12 cm。该仪器具有结构紧凑,不易受干扰,便于操控,速度分辨率高(~0.8%)等特点。低电压加速使得后向的光解碎片离子具有较长的回转时间,即离子牛顿球在TOF轴向上有较大程度的延展,使得相同脉宽的切片能达到更好的效果,可以得到更高分辨的图像。之后又采用稀有气体双光子共振四波混频技术产生可调谐的高分辨真空紫外激光作为仪器的光源,进一步扩大了仪器的研究范围。
2.2 卤代甲烷的光解离研究
在A带,碘代甲烷CX3I分子有两个光解离通道:
CX3I $\xrightarrow{\mathrm{~h} \nu}$ CX3 (v) + I*(2P1/2) ( I* channel )
CX3 (v) + I(2P3/2) ( I channel )
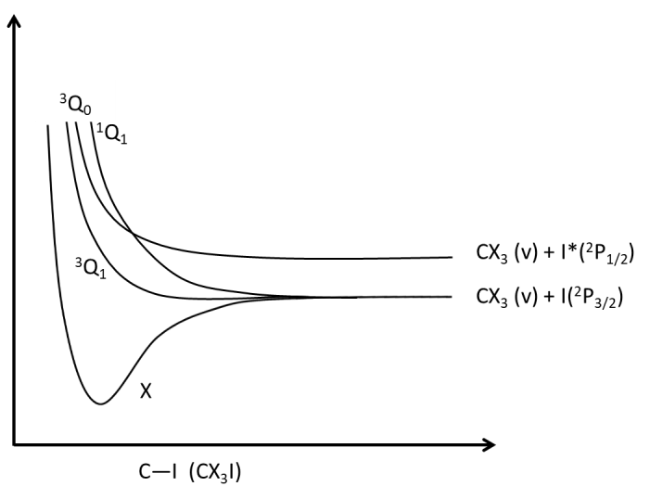
CX3I分子在A带吸收光子后发生σ*$\leftarrow$n跃迁,将导致C-I键的解离。其在A带吸收、解离所涉及的电子态主要有排斥态的3Q1、3Q0和1Q1,且3Q0$\leftarrow$X跃迁为A带最主要的吸收[35]。其中从基态到1Q1和3Q1态吸收为垂直跃迁,对应的直接解离产物为CX3和I(2P3/2),称为基态I通道;从基态到3Q0吸收为平行跃迁,对应的直接解离产物为CX3和I*(2P1/2),称为激发态I*通道。在排斥态势能面3Q0和1Q1之间存在锥形交叉,分子吸收光子初始跃迁到这两个态后,在解离过程中可能会出现势能面间的穿越(Curve crossing,简写为cc,如图4所示)。所以,CX3I在A带光解时存在如下5个光解路径:
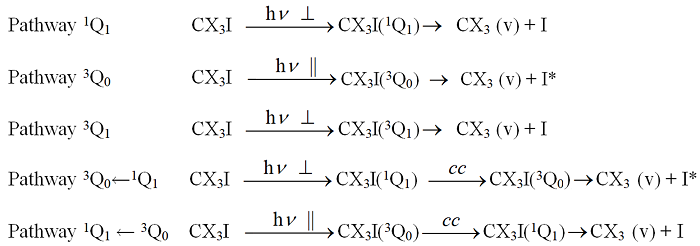
2.2.1 碘甲烷(CH3I)的光解离研究
CH3I是CX3I系列分子中最简单的体系,其光解离过程也最受实验和理论研究关注。CH3基团在母分子中是三角锥型结构,但碎片CH3自由基的平衡构型为平面结构,其伞形振动(Umbrella,ν2)被认为是光解离中最可能且最强烈被激发的振动。因未得到振动态分辨的结果,早期的实验研究对CH3I光解离产生的CH3碎片的振动激发最高布居态一直存在争议。1985年,朱起鹤等[14]利用自主研制的分子束可转动式激光裂解碎片平动能谱仪进行了CH3I在248 nm的光解离实验研究,获得了光解碎片碘原子的飞行时间谱图(图5)。该谱图显示了两个明显的谱峰,分别对应I*和I解离通道,测得了I*量子产率Φ(I*)。经分析,认为在I*通道CH3伞形振动激发最高布居峰在v2 = 2,在I通道CH3伞形振动激发最高布居峰在v2 = 4。实验还测得CH3I分子C-I键的解离能为D0 = 56 ± 1 kcal/mol。在后续的实验中,测得了信噪比更好的实验谱图,获得了光解过程碎片内能激发占可资用能的比,并计算出光解碎片空间角分布的各向异性参数β = 1.85,进一步确认了在248 nm光解时I*和I通道都来自3Q0态平行跃迁的解离[37⇓−39]。1986年,朱起鹤先生访问美国加州大学伯克利分校李远哲教授实验室,重复了CH3I的光解离实验,获得了振动态分辨的光解碎片平动能谱。在实验上确定了248 nm光解时CH3碎片在I*通道振动激发最高布居峰为v2 = 0,在I通道振动激发最高布居峰为v2 = 1;并且还观察到了CH3自由基较弱的C-H对称伸缩振动(ν1)被激发(研究结果未公开发表)。随后的实验和理论研究都支持了这一结论[24,25,40-41]。

利用自行研制的高分辨微型激光光解碎片平动能谱仪,朱起鹤等[42−43]系统开展了CH3I在整个A带的光解离动力学研究。在A带中段及红端的277、280、297和304 nm附近[42],均测得了具有振动分辨的光解碎片平动能谱(图6),获得了一系列光解离动力学参数(表1)。在277~304 nm范围内,在I*通道只观察到了CH3自由基伞形振动的激发,并且其最高布居峰始终保持在v2 = 0。虽然随着解离激光光子能量降低,CH3的内能也随之降低,但内能与可资用能的比Eint/Eavl均约为0.03,基本保持不变。在I通道可资用能远大于I*通道,观察到了相比伞形振动激发较弱的CH对称伸缩振动(ν1)的激发,且最大只到v1 = 1。在较短波长如277.87和279.71 nm时,CH3振动激发最高布居峰在(v1 = 0,v2 = 1)。在298.23和304.67 nm时,CH3振动激发最高布居峰在(v1 = 0,v2 = 0)。在此波段内,当CH对称伸缩振动被激发时,其伞形振动的最高布居峰均为(v1 = 1,v2 = 0)。随着波长变长,可资用能下降,CH3碎片的内能降低,且内能占可资用能的比Eint/Eavl从277 nm附近的0.09下降到304 nm附近的0.06。而总的v1 = 1的布居从277 nm附近的0.20下降到304 nm附近的0.06和0.03。在304.67 nm垂直跃迁(3Q1通道),CH3碎片的内能稍低于对应的平行跃迁(1Q1$\leftarrow$3Q0通道)。通过对光解碎片空间角分布各向异性参数的测量,发现在277~304 nm范围内,I*通道均来自经由3Q0势能面解离的贡献;在解离波长< 280 nm时,I通道均来自经由1Q1$\leftarrow$3Q0解离路径的贡献,当解离波长>280 nm时,来自3Q1的贡献逐渐增大,这和A带吸收光谱的构成一致。在277和304 nm,还测量了各个通道的分支比、I*量子产率Φ(I*)以及势能曲线穿越几率Pcc,如表1所示。根据一维的Landau-Zener公式,计算出1Q1与3Q0势能面锥形交叉点的能量Ec约为32 740 cm−1。
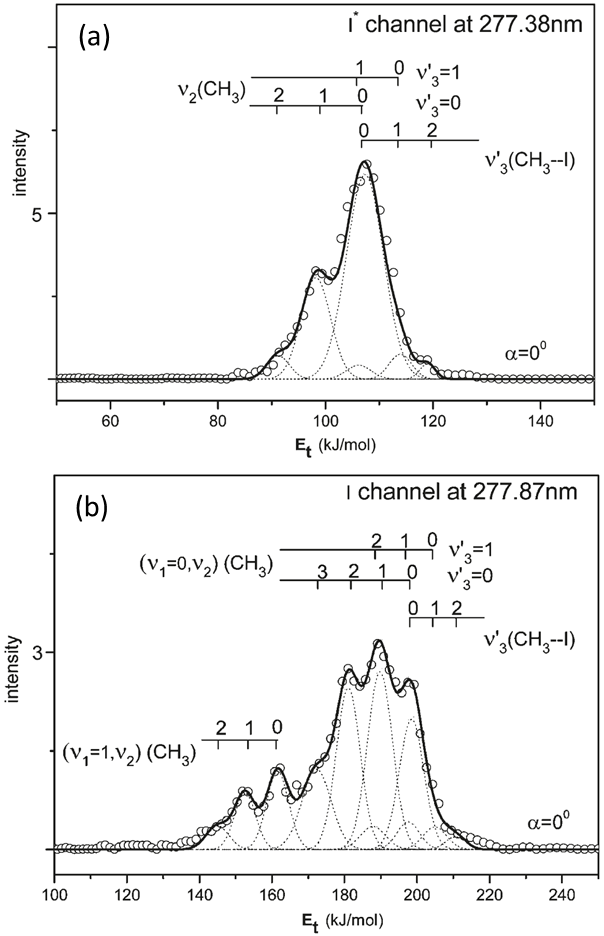
表1 CH3I光解的动力学参数Table 1 The dynamical parameters for the photodissociation of CH3I |
| λ(nm) | Pathway | Channel | Fraction | Φ(I*) | Pcc | Eint/Eavl | β | Ref. |
|---|---|---|---|---|---|---|---|---|
| 225 | 3Q0 | I* | 0.09 | 0.12 | 0.129 | 1.25 | 43 | |
| 3Q0$\leftarrow$1Q1 | I* | 0.03 | 0.08 | 0.104 | ||||
| 1Q1 | I | 0.34 | 0.134 | 0.84 | ||||
| 1Q1$\leftarrow$3Q0 | I | 0.54 | 0.86 | 0.159 | ||||
| 248 | 3Q0 | I* | 0.74 | 0.74 | 0.26 | 0.125 | 1.85 | 14, 37-39 |
| 1Q1$\leftarrow$3Q0 | I | 0.26 | 0.173 | |||||
| 277 | 3Q0 | I* | 0.59 | 0.59 | 0.028 | 1.93 | 42 | |
| 1Q1$\leftarrow$3Q0 | I | 0.41 | 0.41 | 0.087 | 1.91 | |||
| 279.71 | 1Q1$\leftarrow$3Q0 | I | 0.088 | 1.92 | 42 | |||
| 281.73 | 3Q0 | I* | 0.029 | 1.92 | 42 | |||
| 295.91 | 3Q0 | I* | 0.030 | 1.92 | 42 | |||
| 298.23 | 1Q1$\leftarrow$3Q0 | I | 0.065 | 1.70 | 42 | |||
| 304 | 3Q0 | I* | 0.05 | 0.05 | 0.029 | 1.88 | 42 | |
| 1Q1$\leftarrow$3Q0 | I | 0.74 | 0.94 | 0.062 | 1.35 | |||
| 3Q1 | I | 0.21 | 0.057 |
在CH3I吸收A带的蓝端,其光解离表现出与中段和红端显著的不同。朱起鹤等[43]测量的225 nm处光解碎片平动能谱图在I和I*通道均具有较好振动态分辨,通过分析获得了CH3碎片的振动态布居。实验发现不论是在I*还是I通道,CH3碎片的C-H对称伸缩振动均被强烈激发。在I*通道,CH3碎片ν1振动最大被激发到v1 = 1;在I通道,CH3碎片ν1振动最大被激发到v1 = 2。谱图上的振动峰表现出明显的分簇现象,说明CH3的伞形振动不太容易布居到v2 = 4及以上;只有来自1Q1$\leftarrow$3Q0解离路径的I通道没有出现明显的分簇。I*通道的内能占可资用能的比明显低于I通道。还发现初始激发到3Q0态的平行跃迁产生的CH3内能激发要高于初始激发到1Q1态的垂直跃迁,这可能是因为3Q0态的能量比较低,母分子在被激发到3Q0态时CH3基团的伞形和C-H伸缩振动被较强激发,光解离表现出了一定的振动绝热过程。测得4个解离路径3Q0、3Q0 ←1Q1、1Q1、1Q1 ←3Q0的分支比为0.090.030.340.54,I*量子产率Φ(I*) = 0.12,I*通道的β = 1.25,I通道的β = 0.84。实验测得1Q1 ←3Q0的穿越几率Pcc = 0.86,3Q0 ←1Q1的穿越几率Pcc = 0.08,这证明了一维Landau-Zener方程不适合用来预测CH3I在A带蓝端光解时势能面间的穿越几率,因为从一维Landau-Zener方程预测3Q0 ←1Q1和1Q1 ←3Q0的曲线穿越几率Pcc应是相同的。3Q0←1Q1的Pcc = 0.08表明处于1Q1态的CH3I分子很难穿越到3Q0态。
朱起鹤等还观察到了来自热激发导致的C—I伸缩振动(ν3ʹ)被激发的处于振动激发态CH3I母分子的光解离信号。发现来自ν3ʹ振动的母分子光解信号占比从277到304 nm逐渐增强,I通道的热谱效应要低于相应的I*通道;且振动激发态母分子吸收光子后,其1Q1 ←3Q0穿越几率Pcc降低[42]。振动激发态母分子的光解离常常表现出与基态母分子较大的差异[44⇓-46],是态-态反应动力学的重要研究内容,是调控化学反应向着有利方向进行的重要基础。在大气化学和环境化学中,反应物分子处于自然条件下,会有较多的分子布居在振动激发态上,初始振动激发态分子光解离的影响会较显著。为了进一步研究振动激发态CH3I分子的光解离动力学,朱起鹤等采用红外激光预先激发C-H对称伸缩振动到v1ʹ = 1,再研究其在紫外波段的光解离动力学。之前研究人员多研究将断键振动激发对光解离的影响,而朱起鹤等选择其他维度的振动激发,这对研究光解离过程中能量的转移和转化有着重要的意义。
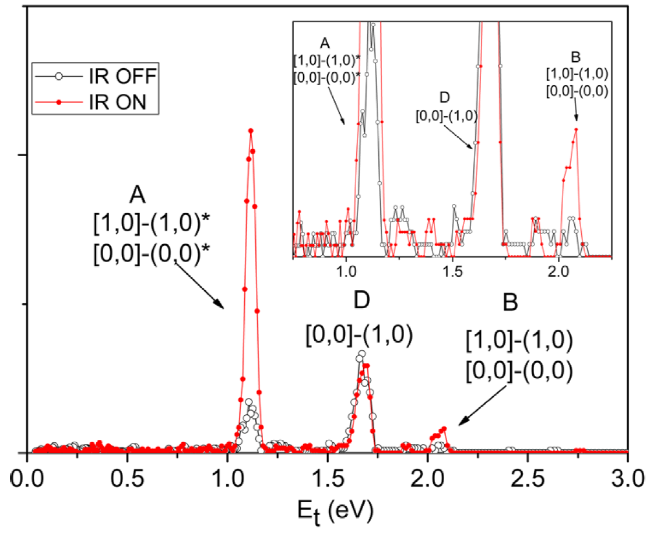
在277 nm处[47],通过探测CH3I(v1ʹ = 1)光解离的碘原子和CH3碎片信号,发现初始v1ʹ = 1的振动激发对CH3I光解离截面的影响较小,初始v1ʹ=1振动绝大部分被保留在CH3碎片中(图7)。CH3(v1 = 1, v2)的伞形振动激发与基态CH3I分子在277 nm附近光解时CH3(v1 = 0, v2)相似;且对比在短波长(225、248 nm)下基态CH3I光解的实验结果,可以清晰地确认CH3碎片的v1 = 1振动激发来源于初始处振动激发保留,而非因为解离能量的升高。此时初始的振动激发在光解中扮演了一个类似于“旁观者”的角色,光解过程可看作是近似振动绝热的。但在304 nm处,CH3I(v1ʹ = 1)的光解与在277 nm处有很大的不同[48]。通过检测光解离过程中产生的碘原子碎片, 发现此时CH3I(v1ʹ = 1)的光解截面与基态碘甲烷光解截面之比约为2.67:1,有很大的增强。光解产物I*的量子产率Φ(I*) = 0.24,远高于基态碘甲烷光解时的Φ(I*);其1Q1←3Q0的穿越几率Pcc = 0.72,低于在此波长下基态碘甲烷光解Pcc值[42,48]。通过探测甲基碎片和碘碎片,确定了CH3I(v1ʹ = 1)光解时产生的甲基碎片CH3(ν1, ν2)的振动激发模式及振动态布居;发现在I通道CH3碎片被激发到v1 = 1的比例要远大于I*通道,说明在I*通道初始母分子的振动激发更难被保留到碎片中。对比在277 nm的光解离数据,发现在低光子能量光解离时,初始母分子的振动激发更难保留到解离碎片中。无论在277还是304 nm处,CH3I(v1ʹ = 1)光解中1Q1←3Q0 的穿越几率Pcc都较基态CH3I分子光解时低,这也导致了其解离产物I*的量子产率Φ(I*)都较高。对于振动激发态CH3I分子的光解离,目前实验和理论研究都还较少,不够系统,尚无法全面、准确地阐释动力学机制,还需开展深入的研究工作。
朱起鹤等还曾研究了碘甲烷二聚体(CH3I)2光解离生成(CH3)2I + I,发现生成的产物碎片平动能非常小,揭示了范德华力在光解过程中起到重要的作用[49]。
2.2.2 全氟代碘甲烷(CF3I)的光解离研究
当CH3I中的H原子被F取代后分子对称性保持不变,CF3I在A带光解离时涉及的势能面和解离路径与CH3I一致。但其解离碎片CF3自由基在基态的平衡构型是三角锥结构,与基态CH3自由基的平面构型相差很大;F原子比H原子要重很多,CF3自由基的振动模式和频率与CH3也有较大的不同。基于Clary[33]与Bowman等[50]的理论计算,本文将CF3自由基频率701 cm−1的振动指认为CF对称伸缩(ν1)模式,频率1086 cm−1的振动指认为CF3碎片的伞形ν2模式(这可能与某些文献的指认相反,请读者加以区分)。Clary[33]和van Veen等[32]通过理论计算预测CF3I在A带光解时,因激发态势能面上存在振动相互作用,CF3碎片会出现明显的双模振动激发(ν1和ν2),但实验研究一直都未能观测到这一现象。
朱起鹤等利用分子束可转动式激光裂解碎片平动能谱仪在248 nm处进行了CF3I的光解研究[15],在I*通道获得了具有振动态分辨的光解碎片平动能谱图。通过对谱图的指认,认为CF3碎片的振动布居最高峰在v1 = 5,并据此计算出C—I键的解离能D0 = 53.8 ± 0.6 kcal/mol,得到了在I*通道光解碎片内能占可资用能的比Eint/Eavl。他们又研究了其在281.73 nm处I*通道的光解碎片平动能谱[51],指认CF3碎片的振动布居最高峰在v1 = 2,其Eint/Eavl约为0.21。谱图上还出现了一个疑似来自CF3碎片双模振动激发的谱峰,但局限于谱图的分辨率和信噪比,尚不能清晰确认。
为了进一步探索CF3碎片双模振动激发,朱起鹤等利用高分辨微型激光光解碎片平动能谱仪在整个A带范围内,系统地开展了CF3I的光解离动力学研究[52⇓-54](表2)。在248 nm处[53],获得了高振动态分辨的I*通道光解碎片平动能谱图(图8),谱峰可明确地分为较强和稍弱交替出现的两组。其中较高谱峰之间的能量间隔ΔEV约为700 cm−1,与CF3自由基的ν1对称伸缩模式的振动频率相符,被指认为CF3碎片的(v1 = 0 ~ 7,v2 = 0)振动态,最高振动布居峰位于(v1 = 4,v2 = 0)。强度稍弱的一组谱峰位于两个(v1,v2 = 0)峰中间的位置,与相邻强峰之间的能量间隔符合(v1,v2 = 1)振动频率差,被指认为CF3碎片的(v1 = 0 ~ 5,v2 = 1)态。这是首次在实验上明确地观察到CF3I光解时碎片CF3自由基双模振动激发。在266和277 nm的I*通道,同样观察到了强弱交替出现的两组振动谱峰,并指认为来自CF3碎片的双模振动激发;最高振动布居峰在266 nm光解时为(v1 = 2,v2 = 0),在277 nm时为(v1 = 1,v2 = 0)。随着波长变长,光子能量降低,v2 = 1振动激发的布居明显降低。在248、266、277 nm光解时,光解过程内能占可资用能的比Eint/Eavl分别为0.220、0.145和0.108,ν2伞形振动激发比∑P(v1, v2 = 1)/∑P(v1, v2)分别为0.41、0.35和0.28,均随着可资用能降低而迅速降低。由于分辨率的限制,不排除CF3碎片ν2伞形振动被激发到更高量子态的可能性。当光解光子能量进一步降低至304 nm附近时[52],在I*通道测得的平动能谱图上有比较多的谱峰,且谱峰的间距很小。经压力实验后发现这些峰来自振动基态或者C-I伸缩振动激发态CF3I(v3ʹ = 0,1,2)分子光解生成CF3碎片的ν1对称伸缩振动激发(v2 = 0),未观察到明确的CF3碎片ν2振动激发。此时CF3碎片的最高振动布居峰为v1 = 1。I*产物空间角分布的各向异性参数β值接近2,表明是经过3Q0势能面直接解离产生。
表2 CF3I光解的动力学参数Table 2 The dynamical parameters for the photodissociation of CF3I |
| λ(nm) | Pathway | Channel | Fraction | Φ(I*) | Pcc | Eint/Eavl | β | Ref. |
|---|---|---|---|---|---|---|---|---|
| 238 | 3Q0 | I* | 0.664 | 0.738 | 0.266 | 1.70 | 54 | |
| 3Q0$\leftarrow$1Q1 | I* | 0.074 | 0.294 | |||||
| 1Q1 | I | 0.178 | -0.04 | |||||
| 1Q1$\leftarrow$3Q0 | I | 0.084 | 0.112 | |||||
| 248 | 3Q0 | I* | 0.220 | 1.85 | 53 | |||
| 266 | 3Q0 | I* | 0.90 | 0.90 | 0.145 | 1.86 | 53 | |
| 1Q1$\leftarrow$3Q0 | I | 0.07 | 0.07 | 0.202 | 1.03 | |||
| 3Q1 | I | 0.03 | ||||||
| 277 | 3Q0 | I* | 0.87 | 0.87 | 0.108 | 1.86 | 53 | |
| 1Q1$\leftarrow$3Q0 | I | 0.086 | 0.09 | 0.154 | 0.98 | |||
| 3Q1 | I | 0.044 | ||||||
| 281.73 | 3Q0 | I* | 0.21 | 51 | ||||
| 304 | 3Q0 | I* | 0.06 | 0.06 | 0.12 | 1.69 | 52, 57 | |
| 1Q1$\leftarrow$3Q0 | I | 0.15 | 0.71 | 0.18 | -0.45 | |||
| 3Q1 | I | 0.79 | 0.15 |
对光解离I通道,在248 nm时未观测到明显信号[53]。这主要是因为248 nm处光子能量远离1Q1和3Q1态吸收中心,CF3I分子无法吸收光子跃迁到这两个态;而激发到3Q0态的分子因为光解离速度较快,通过1Q1←3Q0跃迁解离的几率非常小。在266和277 nm光解的I通道,实验都测量到了具有振动态分辨的光解碎片平动能谱图[53]。谱图揭示了在I通道光解碎片CF3的双模振动也会被同时激发,最高振动峰布居266 nm光解时为(v1 = 5,v2 = 0),在277 nm光解时为(v1 = 3,v2 = 0)。光解碎片内能占可资用能的比Eint/Eavl分别为0.202、0.154,ν2伞形振动激发比∑P(v1, v2 = 1)/∑P(v1, v2)分别为0.47、0.46。这些动力学参数值均高于I*通道,这是因为I通道的可资用能比较大。在304 nm附近,I通道同时存在从3Q1态直接解离(垂直跃迁)和经由1Q1←3Q0解离(平行跃迁)两个解离路径。在两个解离路径上都获得了具有良好振动态分辨的平动能谱图[52],谱图显示CF3碎片的ν1对称伸缩振动被较强激发,未观测到明显的CF3碎片双模振动激发。在这两个解离路径,CF3碎片的最高振动态布居都为v1 = 1;但1Q1←3Q0解离路径内能占可资用能的比Eint/Eavl稍高于3Q1态直接解离,与CH3I在304 nm光解时一致[42]。从266到304 nm光解的I通道,产物空间角分布的各向异性参数β值分别为1.03、0.98和−0.45,表明3Q1态直接解离的产物占比随着光子能量降低而逐渐增多。在277 nm光解时,测得了I*的量子产率Φ(I*) = 0.87,三个解离路径3Q0、1Q1←3Q0、3Q1的分支比为0.870.0860.044,势能面1Q1←3Q0的穿越几率Pcc = 0.09;在304 nm光解时[57],Φ(I*) = 0.06,解离路径3Q0、1Q1←3Q0、3Q1的分支比为0.060.150.79,1Q1←3Q0的穿越几率Pcc = 0.71。

在A吸收带的蓝端,CF3I分子的光解离存在4个路径,即经由3Q0、3Q0←1Q1的I*通道和经由1Q1、1Q1←3Q0的I通道。朱起鹤等在238 nm处进行了CF3I的光解离研究[54],只在3Q0解离路径获得了具有振动态分辨的光解碎片平动能谱图;其他三个路径信号较弱,无法得到较高分辨的谱图。在3Q0解离路径的I*通道,同样观察到了CF3碎片的双模振动激发,其最高振动态布居为(v1 = 6,v2 = 0),Eint/Eavl = 0.266,∑P(v1, v2=1)/∑P(v1, v2) = 0.48。他们测得了4个解离路径的分支比,在I*和I通道产物空间角分布的各向异性参数β值分别为1.70和−0.04。测量得到势能面1Q1←3Q0的穿越几率Pcc = 0.112,而3Q0←1Q1的穿越几率Pcc = 0.294,相差非常大;揭示了在处理锥形交叉这一非常重要的势能面间非绝热相互作用时,需要把穿越方向作为一个重要的考虑项。
2.2.3 氯碘甲烷(I-CH2Cl)的光解离研究
利用高分辨微型激光光解碎片平动能谱仪,他们在277和304 nm处开展了I-CH2Cl分子光解离研究[56],获得了具有清晰振动态分辨的光解碎片平动能谱图(图9)。通过对分辨良好的I*通道谱图的分析,指认在277和304 nm光解时CH2Cl碎片的最大振动布居分别为v = 3和2,并计算出产物具有非常高的转动激发(平均ER/ET ≈ 0.72,转动激发态分别可达J = 95和79),其平动能、转动和振动的能量分配比约为0.48:0.34:0.18。通过谱图上振动峰的能量间距并考虑不同的转动激发,确认光解过程中CH2Cl碎片的C—Cl伸缩振动是最可能被激发的振动模式。在I通道,测量得到产物的转动能和平动能的比例ER/ET略小于I*通道,平动能、转动和振动的能量分配比约为0.45:0.31:0.24。平动能谱上出现非常多的振动峰并被归属于来自CH2Cl碎片的C—Cl伸缩振动激发。不同于I*通道,在I通道平动能谱上的振动峰明显表现出来自两个解离路径的成分(图9),且其贡献随着光子能量的不同而改变。将平动能较小(谱图左边)的成分归属为吸收光子后跃迁到3A'态直接解离生成的I通道,将平动能较大(谱图右边)的成分归属为吸收光子后跃迁到4A'态并经势能面锥形交叉5A'$\leftarrow$4A'后解离生成的I通道。当光子能量从277 nm降低到304 nm时,会有更多的I-CH2Cl分子吸收光子后跃迁到能量较低的3A'态,平动能谱图上平动能较小的成分会有更大的布居。测量发现I*和I通道碎片空间角分布各向异性参数β值都接近2,表明两个反应通道主要都是经过平行跃迁激发的。测得在277和304 nm光解时I*通道量子产率Φ(I*)分别为0.36和0.26。当光解的光子能量降低时,激发态分子通过势能交叉区域的速度降低,时间变长,更多的激发态分子将从4A'穿越到5A'生成基态I,与此同时,会有更多的分子被激发到直接生成基态I通道的3A';这些都会导致Ф(I*)降低。双卤代甲烷的光解与CX3I光解相比,呈现显著的不同,目前的研究尚未系统地揭示其光解动力学机制,还需要实验与理论相结合进一步地深入探索。

2.3 卤代乙烷的光解离研究
碘乙烷(C2H5I)是最简单的碘代烷烃之一,它在A带光解离涉及的激发态势能曲线类似于CH3I。Wilson等[18]曾用分子束激光裂解产物谱仪研究过C2H5I的光解离,但由于仪器分辨率不高,在TOF谱上未能分辨出激发态碘I*和基态碘I通道。朱起鹤等利用自行研制的分子束可转动式激光裂解碎片平动能谱仪研究了C2H5I在248 nm的光解[38-39,58],实现了I*和I通道在TOF谱上的分离(图10),测得I*通道量子产率Φ(I*)为0.70(表3)。研究发现,两个光解离通道都遵从冲击模型(Impulse model)机制,它们的解离是发生在沿着C—I键排斥态势能面上的快速过程,即C—I键吸收光能后,其能量来不及在分子的其他自由度中作随机分布,就发生了解离。根据碎片TOF谱和能量守恒,得到了两个光解通道中C2H5自由基的内能分布(表3)。在I*通道,除了伞形振动(540 cm-1)被激发外,C—C键的伸缩振动(1138 cm-1)很可能也被激发;在I通道,C2H5自由基中除了上述振动外,属于反对称振动模式的扭振动(193 cm-1)和摆振动(1175 cm-1)也很可能被激发。
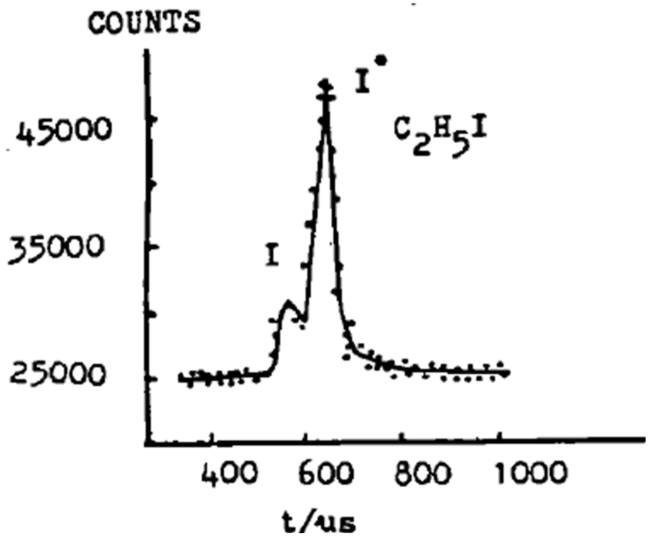
表3 248 nm下部分碘代烃光解离的动力学参数Table 3 The dynamical parameters for the photodissociation of partial iodohydrocarbon at 248 nm |
| Reaction Channels | Φ(I*) | Eint(R)/Eavl | Ref. |
|---|---|---|---|
| C2H5I→C2H5 + I* | 0.70 | 0.32 | 38 |
| C2H5 + I | 0.39 | ||
| n-C3H7I→n-C3H7 + I* | 0.62 | 0.49 | 38 |
| n-C3H7 + I | 0.54 | ||
| i-C3H7I→i-C3H7 + I* | 0.49 | 0.63 | 38 |
| i-C3H7 + I | 0.64 | ||
| n-C4H9I→n-C4H9 + I* | 0.31 | 0.70 | 39 |
| n-C4H9 + I | 0.74 | ||
| t-C4H9I→t-C4H9 + I | 0.76 | 39 | |
| n-C5H11I→n-C5H11 + I* | 0.67 | 0.77 | 59 |
| n-C5H11 + I | 0.76 | ||
| CH2=CHI→CH2=CH + I* | 0.57 | 0.31 | 60 |
| CH2=CH + I | 0.41 |
后来,朱起鹤等[61]又研究了C2H5I在280和304 nm附近的光解动力学,均获得了部分振动态分辨的平动能谱图(图11),谱图上的振动峰被归属为碎片C2H5自由基的伞形振动模式(540 cm-1)的激发。还测得C2H5I中C—I键的解离能D0 = 53.4 ± 0.7 kcal/mol。在281.73和 304.02 nm处I*通道,内能占可资用能的比Eint/Eavl分别为0.221和0.224;在279.71和304.67 nm处I通道,Eint/Eavl分别为0.252和0.259(表4)。对比248 nm的光解离结果可以发现,当可资用能减小时,分配到解离产物乙基碎片内能的比例Eint/Eavl会更小。
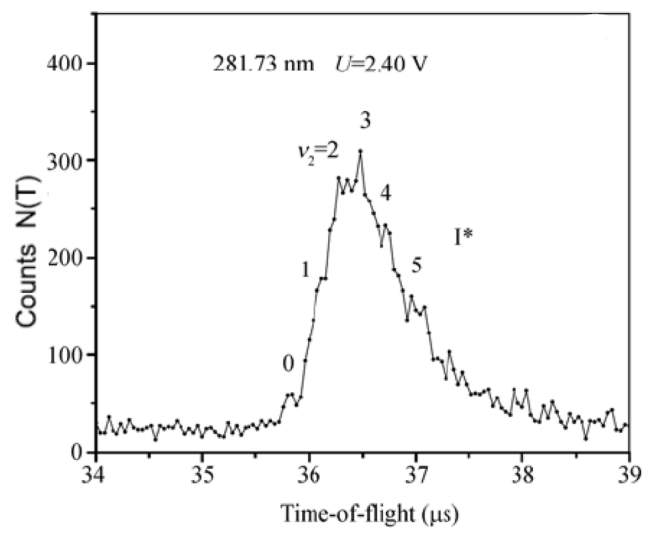
表4 279’305 nm范围部分碘代烃光解的动力学参数Table 4 The dynamical parameters for the photodissociation of partial iodohydrocarbon at 279~305 nm |
| λ/nm | Channels | Eint/Eavl | β | Ref |
|---|---|---|---|---|
| 281.73 | C2H5 + I* | 0.221 | - | 61 |
| 304.02 | 0.224 | - | ||
| 279.71 | C2H5 + I | 0.252 | - | 61 |
| 304.67 | 0.259 | - | ||
| 281.73 | C2F5 + I* | 0.52 | 1.70 | 16 |
| 304.02 | 0.50 | 1.64 | ||
| 279.71 | C2F5 + I | 0.60 | 1.25 | 16 |
| 304.67 | 0.55 | 0.88 | ||
| 281.73 | n-C3H7 + I* | 0.48 | 1.68 | 62 |
| 304.02 | 0.49 | ’2.00 | ||
| 279.71 | n-C3H7 + I | 0.52 | ’2.00 | 62 |
| 304.67 | 0.52 | 1.57 | ||
| 281.73 304.02 | i-C3H7 + I* | 0.61 | 1.72 | 62 |
| 0.65 | 1.75 | |||
| 279.71 | i-C3H7 + I | 0.62 | 1.32 | 62 |
| 304.67 | 0.49 | 1.31 |
相比C2H5I,全氟代碘乙烷(C2F5I)的研究相对较少,朱起鹤等[16]利用高分辨微型激光光解碎片平动能谱仪研究了C2F5I在280和304 nm附近的光解离。在I*通道,得到了C2F5自由基具有振动态分辨的光解碎片平动能谱(图12),并将振动峰归属为CF2 wag模式的激发(ν11 = 366 cm-1)。在281.73和304.02 nm光解离I*通道,测得Eint/Eavl分别为0.52和0.50;在279.71和304.67 nm光解离I通道,Eint/Eavl分别为0.60和0.55(表4)。C2F5I在光解离过程中C2F5碎片的内能激发非常高,且远高于C2H5I,与CF3I光解离相较于CH3I的结论一致[42,52-53]。这说明F原子取代H原子后,碘代烃光解离时会有更多的可资用能被分配到烃基碎片当中。在I*通道得到光解碎片空间角分布各向异性参数β值均接近2,说明I*通道主要来自3Q0态解离的贡献。而在279.71和304.67 nm的I通道测量得到β = 1.25和0.88(表4),说明经由3Q1态直接解离和经由1Q1←3Q0解离两个路径均对I通道有贡献;且随着解离波长的增长,经由3Q1直接解离所占比例成分越来越大,但在这一波段内1Q1←3Q0通道的贡献始终占主导。

2.4 其他卤代烃的光解离研究
朱起鹤等[38,63]用分子束可转动式激光裂解碎片平动能谱仪在248 nm处研究了碘代丙烷(C3H7I)的光解离,获得的谱图清晰分辨出碘代正丙烷(n-C3H7I)和碘代异丙烷(i-C3H7I)光解时I*和I两个光解离通道,测量了I*的量子产率Φ(I*)分别为0.62和0.49,还获得了碎片内能占可资用能比Eint/Eavl(表3)。他们还研究了n-C3H7I和i-C3H7I在280和304 nm附近的光解离行为[62],在I*和I通道均得到了具有振动态部分分辨的平动能谱。谱图显示在n-C3H7I光解离过程中碎片的RCH2变形振动(频率530 cm−1)最容易被激发,而i-C3H7I光解离过程中碎片的HC(CH3)2面外弯曲振动(频率364 cm−1)是易被激发的振动模式。实验中测得了n-C3H7I和i-C3H7I光解离后可资用能的分配(表4),发现其与248 nm光解时Eint/Eavl相接近。相较于n-C3H7I,i-C3H7I光解离时会有更高比例的内能分配。在此波长范围内n-C3H7I光解离时在I和I*通道测得的β值均接近2,说明两个光解通道均来源于1Q1←3Q0及3Q0解离路径。i-C3H7I光解离时I*通道的β值接近2,说明主要来源于3Q0直接解离;但在I通道的β值较小,说明有部分I通道信号来自3Q1直接解离的贡献。
利用分子束可转动式激光裂解碎片平动能谱仪,朱起鹤等研究了更大尺寸的碘代正丁烷(n-C4H9I)、碘代叔丁烷(t-C4H9I)[39]及碘代正戊烷(n-C5H11I)[59]分子在248 nm下的光解离机理。他们测得n-C4H9I和n-C5H11I光解离时I*通道的量子产率Φ(I*)分别为0.31和0.67,获得了解离碎片的内能占可资用能比(表3)。所获得的碘代烷烃光解离实验结果表明,随着烷烃基团的增大,光解离过程中更大比例的可资用能将转变成烷基的内能。这主要是因为烷基的尺寸增大,碎片振动自由度增多,内能被激发的将更多。碘代烷烃分子光解离I*通道量子产率Φ(I*)和各解离路径分支比,主要由吸收光子时从分子基态到3Q0、1Q1和3Q1态的跃迁强度以及解离过程中3Q0和1Q1态双向势能曲线的穿越几率控制,而激发态碘代烷烃分子越过交叉区的速度和穿越方向是影响穿越几率的重要因素。
除了碘代烷烃外,他们还利用分子束可转动式激光裂解碎片平动能谱仪,研究了碘代乙烯(CH2=CHI)在248 nm的光解离[60]。同碘代烷烃的光解类似,CH2=CHI的光解离有两个通道I*(2P1/2)和I(2P3/2)。实验测得I*通道的量子产率Φ(I*) = 0.57,C-I键的键解离能D0 = 61.9 ± 1.0 kcal/mol,获得了解离过程中的能量分配(表3)。研究结果证明了CH2=CHI的光解离是发生在沿着C—I键的排斥态势能面上的快速过程。在I*通道,CH2=CH碎片的弯曲和面外弯曲振动模式ν7(918 cm-1),ν9(783 cm-1)和ν8(827 cm-1)最有可能被激发;在基态I通道,除了以上模式激发外,C—C伸缩振动模式(1670 cm-1)也可能被激发。
他们还研究了溴代丙烯中3-C3H5Br和1-C3H5Br在193nm的光解离动力学[64]。根据实验得到的Br+飞行时间谱和能量守恒分析发现,3-C3H5Br和1-C3H5Br的解离不同于碘代烷烃的解离,不能用冲击模型来解释,它们的解离与氢转移导致的异构化之间存在竞争。
朱起鹤团队系统地研究了卤代烃等分子在A带的光解离,得到了其光解规律,提出了它们的微观反应机理,为治理大气污染、保护臭氧层等方面提供了理论依据。
3 重要小分子的光电离动力学研究
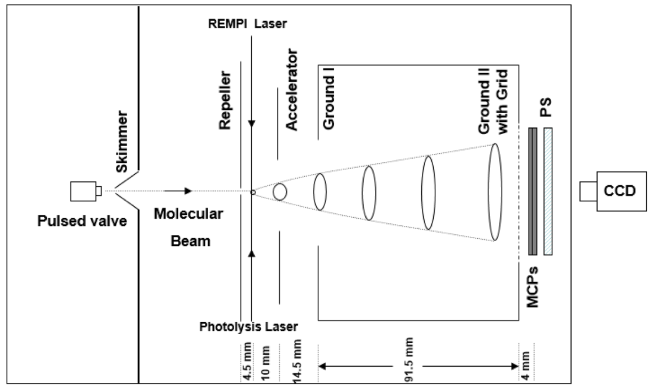
3.1 REMPI/MATI和IR-UV光谱实验装置
朱起鹤团队自主搭建了基于直线式飞行时间质谱仪的共振增强多光子电离光谱和质量解析阈值电离光谱实验装置。该装置主要由以下几个部分组成:内部腔体系统(束源室、电离室、自由飞行区和离子探测区)、真空系统、激光系统、时序控制系统和信号采集系统(图13)。后来又对仪器进行了改进,采用激光脱附进样,实现了对高沸点、低热稳定性分子的REMPI/MATI光谱研究。几种共振双光子电离(R2PI)光谱和MATI光谱的原理示意图如图14所示。又引入红外激光,实现了几种红外-紫外双共振IR/UV光谱技术,包括:研究单体和团簇在电子基态(S0态)振动的IR-UV光谱、研究单体在离子电子基态(D0态)振动的自电离检测的红外光谱(ADIR),以及研究团簇在D0态振动的红外光解光谱(IRPD)(图15)。以这些实验装置为基础并结合理论计算,朱起鹤团队开展了一系列苯衍生物分子的光电离光谱研究。本文介绍的一些重要苯衍生物分子从S0态到第一电子激发态(S1态)的跃迁能(E1)和电离能(IE)等重要光谱数据如表5所示。
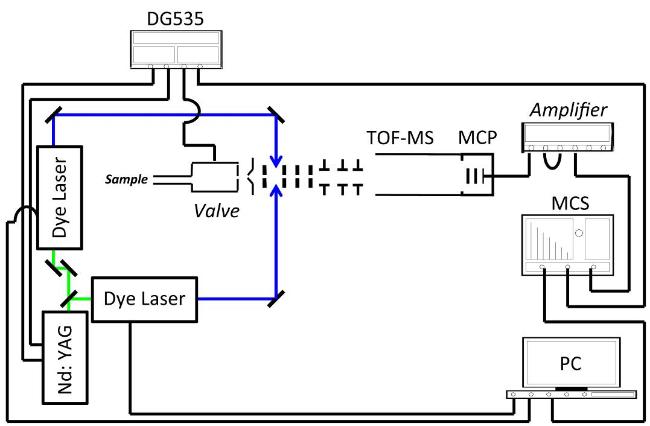
图13 REMPI/MATI光谱实验装置示意图Fig. 13 The schematic diagram of REMPI/MATI spectroscopy experimental device |
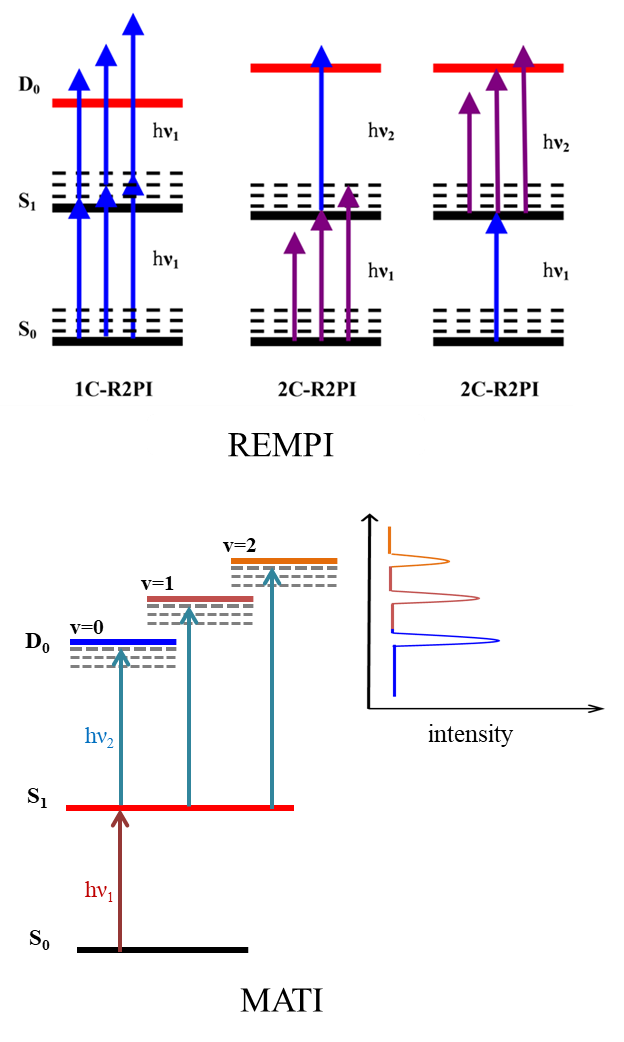
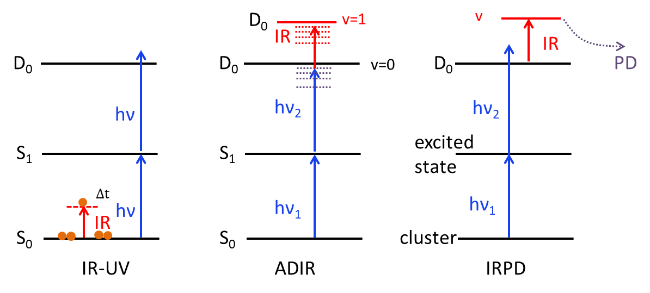
图15 IR-UV、ADIR和IRPD光谱原理示意图Fig. 15 The principles of IR-UV, ADIR, and IRPD spectroscopy |
表5 若干典型苯衍生物分子的第一电子激发态跃迁能(E1)和电离能(IE)汇总表Table 5 Summary of electronic transition energies (E1) and ionization energies (IE) for several typical benzene derivative molecules |
| Molecules | E1 (cm−1) | IE (cm−1) | Ref. |
|---|---|---|---|
| cis m-fluorostyrene | 34403 | - | 65 |
| trans m-fluorostyrene | 34663 | - | 65 |
| p-methylstyrene | 34276 | - | 66 |
| p-fluoroanisole | 35149 | - | 67 |
| cis p-methoxystyrene | 33242 | - | 68 |
| trans p-methoxystyrene | 33324 | - | 68 |
| p-chloroanisole | 34859 | - | 69 |
| cis 3-chloro-4-fluoroanisole | 34703 | 67349 | 70 |
| trans 3-chloro-4-fluoroanisole | 34747 | 67595 | 70 |
| cis 3-chlorostyrene | 33766 | 69701 | 71 |
| trans 3-chlorostyrene | 34061 | 69571 | 71 |
| cis m-aminostyrene | 30937 | 61278 | 72 |
| trans m-aminostyrene | 31140 | 61495 | 72 |
| 3,5-difluoroanisole | 37595 | 70096 | 73 |
| cis 3-chloro-5-fluoroanisole | 36468 | 69720 | 74 |
| trans 3-chloro-5-fluoroanisole | 36351 | 69636 | 74 |
| cis 3-fluoro-N-methylaniline | 33816 | 61742 | 75, 76 |
| trans 3-fluoro-N-methylaniline | 34023 | 61602 | 75, 76 |
| cis 4-chloro-3-fluoroanisole | 35443 | 67585 | 77 |
| trans 4-chloro-3-fluoroanisole | 35326 | 67324 | 77 |
| trans 2-fluoro-N-methylaniline | 34010 | 61101 | 78 |
| cis 3-chloro-N-methylaniline | 33003 | 61531 | 79 |
| trans 3-chloro-N-methylaniline | 32886 | 61625 | 79 |
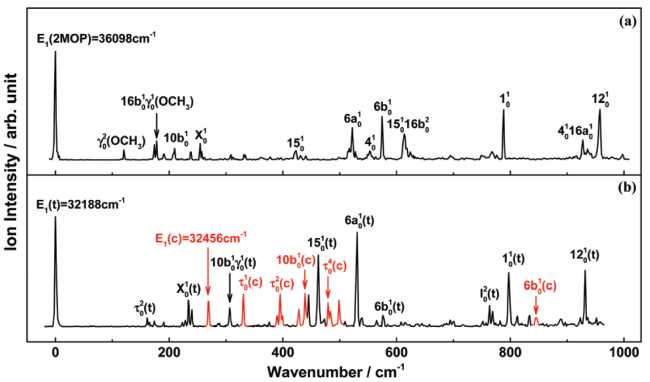
| cis 2-methoxypyridine | 36098 | 69379 | 80 |
| cis 2-N-methylaminopyridine | 32456 | 62518 | 80 |
| trans 2-N-methylaminopyridine | 32188 | 62709 | 80 |
3.2 苯衍生物的REMPI和MATI光谱研究
苯的衍生物是许多医药品、工农业产品和新材料的合成单元,也是环境化学和生物化学领域的重要研究对象。这些化合物的电子态和离子态的光谱特征引起越来越多研究工作者的关注。相对电子基态,分子在电子激发态和离子态的光谱研究相对较少,尤其是分子较大、原子数较多的苯衍生物体系。
应用1C-R2PI光谱技术研究了对氟苯甲醚[67]在S1态的振动光谱,结合量子化学理论计算对分子结构进行了分析,对激发态光谱进行了指认和解析,并分析了不同取代基在S1态的取代基效应。测得对氟苯甲醚的E1值为35 149 cm-1,相比苯甲醚E1红移了1234 cm-1。结合理论计算,对实验获得的对氟苯甲醚在S1态的活性振动模式进行了指认和模拟。研究发现在S1态,由于p-π共轭程度更高,连接氧原子与苯环的C—O键缩短,具有了部分双键的性质。通过与苯衍生物及其对应的对位氟取代化合物跃迁能的比较,发现对位氟取代引起S1态的能量降低要大于电子基态和离子基态的能量降低。因此从基态跃迁到S1态,跃迁能发生了红移;而从S1态跃迁到离子基态,跃迁能则发生了蓝移。
通过理论计算发现对甲氧基苯乙烯[68]的分子骨架在基态和激发态都是平面构型,存在顺式(cis)和反式(trans)两种稳定的旋转异构体。无论是S0还是S1态,反式异构体都较顺式稳定。在S1态,取代基与苯环的作用比在S0态更强,而且乙烯基与苯环的作用要强于甲氧基与苯环的作用;C4—O键和C1—Cα键具有部分双键的性质,而Cα=Cβ键的键长则随着C1—Cα共轭程度加强而拉长了。由1C-R2PI光谱获得顺式和反式异构体的E1值分别是33 242和33 324 cm−1。结合理论计算对光谱实验获得的两个异构体在S1态的振动模式进行了指认。
利用双色共振双光子电离(2C-R2PI)光谱实验结合理论计算,他们研究了取代基效应、异构体效应和同位素效应对3-氯-4-氟苯甲醚(3C4FA)[70]分子性质的影响。通过1C-R2PI和2C-R2PI光谱测量,得到顺式3C4FA的E1和IE分别为34 703和67 349 cm−1;而反式3C4FA的E1和IE分别为34 747和67 595 cm−1。结合理论计算,对3C4FA的氯同位素取代和顺反异构体的R2PI光谱峰进行指认,发现异构体效应对3C4FA的跃迁能、电离能及振动频率的影响比同位素效应的影响大。
针对取代基效应、构象异构体效应对间氨基苯乙烯[72]分子性质的研究中,发现顺式和反式间氨基苯乙烯的E1分别为30 937和 31 141 cm-1,相对于对氨基苯乙烯的E1发生了红移;而这与大多数间、对位双取代苯衍生物分子第一电子激发态能的顺序相反。光谱上的活性振动模式主要涉及分子在S1态时苯环内的平面振动,且取代基的相对位置对振动频率有影响。实验测得顺式和反式间氨基苯乙烯的IE分别为61 278和61 495 cm−1,并获得顺式和反式异构体在S0、S1和D0态的能差分别为30、234和247 cm-1,发现顺式异构体在S0、S1和D0态都比反式异构体稳定。
对于间氯苯乙烯[71],他们利用REMPI和MATI光谱测得顺式和反式间氯苯乙烯的E1分别为33 766和34 061 cm-1,绝热电离能分别为69 701和69 571 cm-1,获得顺式和反式异构体在S0、S1和D0态的能差分别为218、513和88 cm-1,发现顺式异构体在S0、S1和D0态都比反式异构体稳定。在REMPI和MATI光谱上观察到的振动模式主要涉及苯环平面的振动和与取代基有关的振动。
采用REMPI光谱实验和理论计算研究了cis和trans 3-氯-5-氟-苯甲醚(3C5FA)[74]在不同电子态的分子构型和激发态的振动光谱。实验发现cis-和trans-3C5FA的E1值分别为36 468和36 351 cm-1。通过分析光谱数据发现,构象异构效应对S1态某些振动模式频率的影响要比同位素效应大,而后者的影响主要取决于振动模式中Cl原子的参与程度。他们利用光电子效率(PIE)实验方法,测得cis和trans-3C5FA的电离能分别为69 720和69 636 cm-1。他们还发现双卤素取代对3C5FA的E1和电离能产生的累积效应相较于前人总结的多取代基加和性规律有一定偏差,但这一加和规律依然对预测复杂的多取代衍生物的E1和电离能具有一定的参考价值。
对3,5-二氟苯甲醚(3,5-DFA)及其Ar团簇[73]的光电离光谱进行了研究。实验测得3,5-DFA分子的E1值为37 595 cm−1,相比顺式间氟苯甲醚、反式间氟苯甲醚和苯甲醚的E1分别蓝移933、776和1212 cm-1;3,5-DFA的电离能为70 096 cm-1。发现3,5-DFA 相比苯甲醚的E1和电离能蓝移实验值要比依据加和规律得到的蓝移预测值大497和324 cm-1。通过理论计算,发现在3,5-DFA···Ar团簇中Ar原子位于苯环平面上方,且偏向于OCH3基一侧。这一发现证明Ar原子与3,5-DFA以较弱的色散作用结合,Ar对3,5-DFA分子本身性质的影响很小,也解释了相较与单体其团簇的E1值仅红移9 cm−1。
3.3 苯衍生物分子的构象异构研究
构象异构(Conformational isomerism)是指有机物分子由于单键的旋转而使得分子各原子或原子团在空间产生不同排列方式的一种立体异构现象。构象异构现象在生物体系广泛存在,并起着至关重要的作用。常见的苯衍生物如苯甲醚、苯酚、苯乙烯、苯胺、N-甲基苯胺等,由于C—O、C—C和C—N单键可以旋转,这些分子的衍生物往往也存在构象异构现象。可以通过理论计算和光谱数据来推测不同构象体的相对稳定性。
朱起鹤等研究了以苯甲醚和N-甲基苯胺衍生物为代表的苯衍生物的构象异构现象。理论计算表明2-氟-N-甲基苯胺(2FNMA)[78]、3-氯-N-甲基苯胺(3ClNMA)[79]和4-氯-3-氟苯甲醚(4Cl3FA)[77]在S0、S1和D0态均存在cis和trans两种构象。但在实验上只观测到trans-2FNMA构象的光电离光谱,通过理论计算获得了两种不同构象的势能曲线;而对3ClNMA和4Cl3FA均观测到了cis和trans构象的光电离光谱。通过REMPI和 MATI光谱测得了这些分子各构象的E1和IE的精确数据,获得了其S1和D0态的振动特征;进一步结合理论计算,总结了取代基效应和构象异构效应对这些分子在基态、激发态和离子态性质的影响。

3.4 苯衍生物及其团簇的IR/UV激光光谱
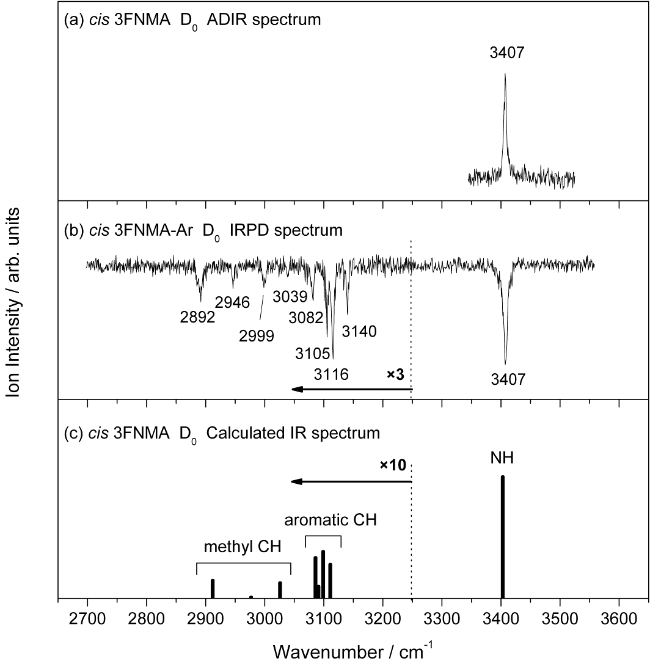
朱起鹤等利用REMPI/MATI光谱结合IR-UV、ADIR、IRPD等方法,研究了3-氟-N-甲基苯胺(3FNMA)及3FNMA···Ar1团簇的光谱,获得了这些体系在S0和D0态的光谱信息[76],分析了取代基效应和构象异构体效应对3FNMA不同振动模式光谱行为的影响,并采用共轭效应和重杂化效应对其进行了解释。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
通过比较cis和trans异构体的IR光谱实验结果,发现3FNMA的CH伸缩振动表现出一定的异构体效应,且在D0态时的异构体效应相比在S0态时减小。两个异构体的CH伸缩振动光谱均因费米共振效应而存在不同程度的谱峰裂分。通过比较3FNMA在S0和D0态的IR光谱,发现电离对NH和CH伸缩振动产生的影响是明显不同的。前者在D0态的振频红移(~80 cm-1),IR吸收强度增大;而后者在D0态的振频蓝移(30~60 cm-1),IR吸收强度显著降低。3FNMA分子光谱特征表明这种分子内部存在共轭效应和重杂化效应。
朱起鹤团队采用IR/UV激光光谱技术结合REMPI和MATI光谱方法,在实验上获得了分子在不同电子态的更全面的光谱信息,并将研究对象扩展至团簇,为深入探索团簇构型和分子间作用等重要科学问题提供强有力的实验方法。
4 总结与展望
朱起鹤先生是我国分子反应动力学研究领域的开创者之一。本文仅从重要小分子的光解离和光电离微观动力学领域回顾了朱起鹤先生的研究工作。朱起鹤先生在科学仪器研制方面独居匠心。他善于利用创新性的原理,将复杂的科学仪器小型化、简单化,能够在提高仪器性能的同时降低成本,并使其具备易于操作和维护等特点。这不仅为科学仪器的研制与创新开辟了新的思路,也为培养科学仪器研制人才提供了宝贵的启示。
朱起鹤先生及其团队深耕于卤代烃光解离动力学,系统地研究了一系列卤代烃的光解离过程,获得了其光解离规律,并提出了微观反应机理,促进了分子光解离动力学理论的发展,还为大气污染治理和环境保护等领域提供了重要模型参数和理论依据。他们的研究也表明卤代烃的光解离动力学研究非常重要,其反应机理非常复杂,值得深入研究。
从光解离机理角度来看,仍有待将研究推进到超快时间分辨动力学、量子态选择激发态解离动力学。首先,势能面的锥形交叉广泛存在于卤代烃的光解离过程中,是影响分子光解离动力学的核心要素之一。然而,当前多数研究仍依赖于通过实验测量反应产物并结合理论计算的方式,间接地推测势能面锥形交叉区域的相关信息。值得庆幸的是,阿秒激光、超快电子衍射等技术的出现,使直接探测锥形交叉区域的动力学过程成为可能[82-83]。将这些超快实验技术应用于卤代烃光解离研究中,深入探索势能面间的锥形交叉,对于揭示分子光解离的物理化学本质以及理解电子态间复杂的非绝热相互作用具有举足轻重的意义。其次,目前卤代烃的光解离研究主要关注基态分子,在态-态层次上深入研究处于振动激发态母分子光解离的工作还非常缺乏。采用红外激光等方式选择性激发母分子的特定振转模式,再进一步研究其光解离[47-48],可以获得不同振转模式激发母分子的光解截面信息,了解不同振转模式与光解坐标间的耦合作用,阐述母分子的初始振转与光解产物内能激发的联系,得到不同振转模式对光解产物量子产率的影响,有助于真正实现从量子态层次上深入理解化学反应的机理。
从卤代烃光解离实际应用模型来看,仍需对更多的分子在更宽波段进行系统研究,最终建立完备的数据库,从而在工业生产和大气环境领域发挥重要作用。对于基态碘代甲烷在A带的光解离,无论是实验还是理论研究都已经比较充分,能够很好地阐述其微观反应机制,但在更高吸收带光解离的实验和理论研究都还比较缺乏。而到达地球大气层的太阳光辐射在短波长处也有着较强的分布,其对污染物分子的解离和反应都有着重要作用。但目前相关的研究数据尚不充足。对于更复杂卤代烃的光解离,目前的实验和理论研究都还不系统。因此,更系统、全面地实验结合理论研究多种卤代烃在整个太阳光紫外辐射波段内的光解离动力学,将有助于建立精确而完善的模型来评估对地球大气和环境的影响[2-3]。
在重要小分子光电离光谱的研究方面,朱起鹤先生及其团队通过对一系列苯衍生物分子及其团簇的激发态和离子态振动光谱的实验和理论研究,获得了不同电子态下分子的几何结构、振动频率、激发能以及电离能等方面的详细信息,深入探究了取代基效应、构象异构体效应、同位素效应等对分子和团簇光谱行为的影响和规律。通过对这类苯衍生物模型分子的研究,有助于更好地理解芳香族化合物或功能材料分子等复杂体系的结构及其物理化学特性,对探索生物分子功能、提出新的光化学反应机理、开发和改进新型功能材料等许多领域都具有重要的参考价值。在这一方向继续深入研究,并加以拓展,有望取得重要突破。
从实验技术角度看,对于含有多个杂原子的杂环、复杂侧链以及多取代基取代的芳香化合物尤其是生物分子,因其质量数较大、沸点较高或热稳定性不好,对这类分子的激发态和离子态光谱研究还存在挑战。首先需要改进实验进样系统,例如可采用激光脱附等技术,在不破坏样品分子的前提下增加进样量,以获得较高的样品分子浓度进行后续光谱研究;其次需提高质谱分辨率和探测灵敏度,以实现对高质量数样品的精确光电离光谱的研究[84-85]。对于激发态寿命短、电子激发跃迁Franck-Condon因子较低等导致光电离谱信号强度较弱的体系,可采用诸如皮秒激光等短脉冲光源、真空紫外激光等高光子能量光源直接电离等技术来获得这些分子的光电离机理[84⇓-86]。从应用角度来看,随着技术和理论的不断发展,光电离光谱将在环境监测领域、材料、生物医药等发挥更加重要的作用。例如,光电离光谱技术可用于实时监测大气、水质等环境中的污染物,为环境保护提供重要数据支持;实时原位测量生物分子在光电离过程中的光谱特征,可以揭示其分子结构和相互作用机制。此外,人工智能技术的快速发展为光电离光谱领域的研究提供了强有力的发展助力。例如,通过训练算法处理大规模的光谱数据,可以大幅提高光谱分析的效率和准确性,帮助研究人员更好地理解复杂反应体系的光谱信息。
小分子的光解离和光电离研究领域具有广阔的发展前景和重要的应用价值,需要科研人员不断深入探索和研究。对分子光解离和光电离过程微观物理化学机制的深刻认识,将为实现化学反应调控这一“终极目标”起到重要作用。