



1 引言
2 电池高电压失效机制
2.1 电解液分解
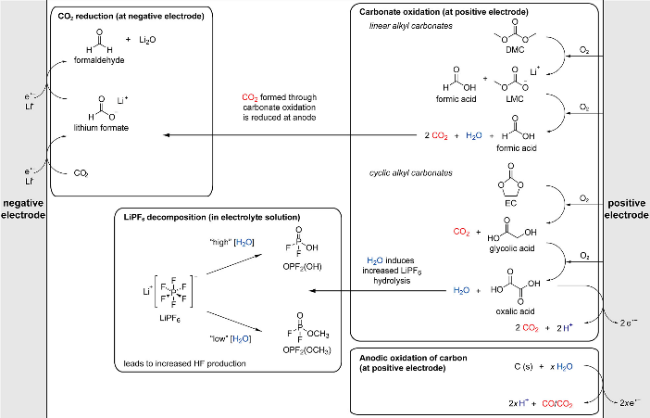
2.2 过渡金属离子溶出
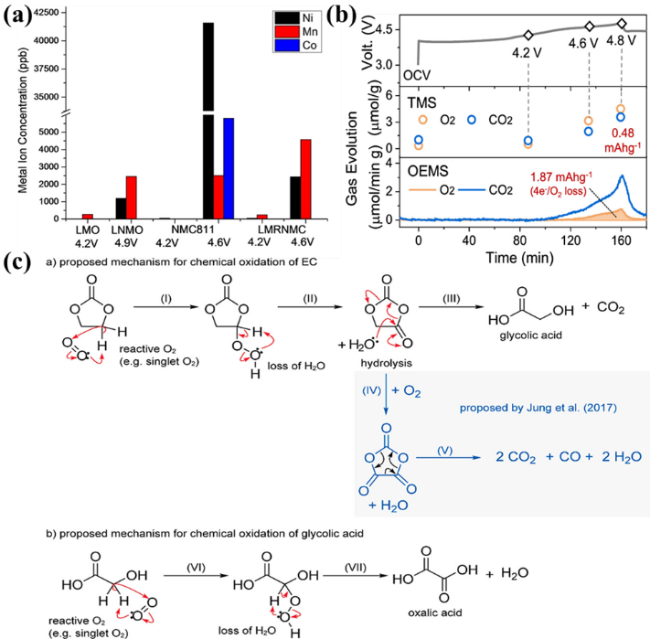
图2 (a)不同阴极电池100圈循环后石墨阳极表面过渡金属离子含量[12];(b)LiCoO2在不同电压下的滴定质谱和在线电化学质谱[21];(c)EC的化学氧化机理[8]Fig. 2 (a) Transition metal ion content on the surface of graphite anode after 100 cycles of different cathode batteries[12]. (b) TMS and OEMS results for LiCoO2[21]. (c) Proposed Mechanism for the Chemical Oxidation of Ethylene Carbonate[8] |
2.3 HF侵蚀
3 高电压电解液的研究进展
3.1 提升电解液本征稳定性的研究进展
3.1.1 高氧化稳定性溶剂
3.1.1.1 含氟溶剂
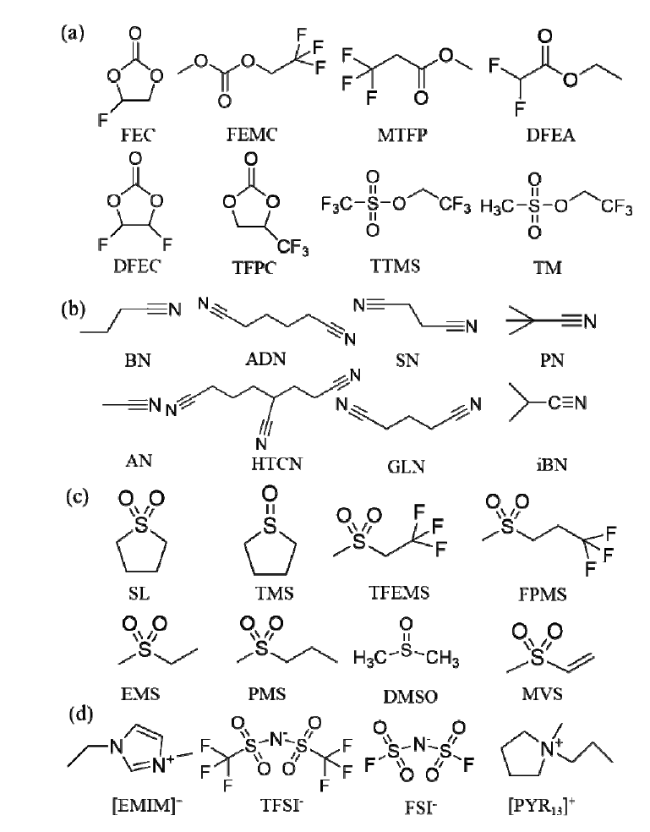
图3 常用(a)含氟溶剂;(b)腈类溶剂;(c)砜类溶剂;(d)离子液体的分子结构Fig. 3 The molecular structures of some (a) fluorinated solvents; (b) nitrile-based solvents; (c) sulfone-based solvents; (d) ionic-liquid solvents |
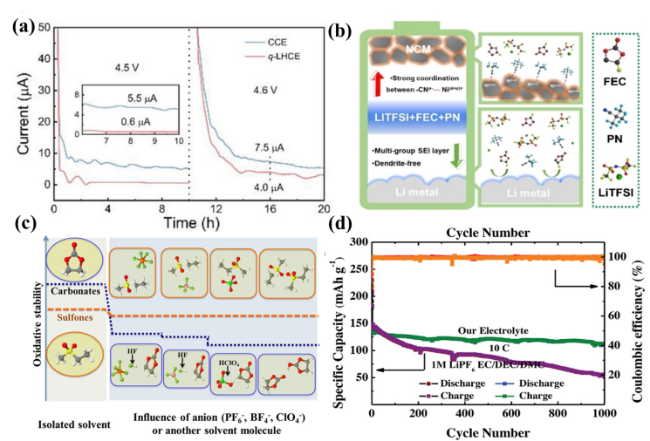
图4 (a) 使用不同电解液的Li/LCO电池在4.5和4.6 V恒压下的漏电流[23];(b) TFP电解液在电池中的作用示意图[42];(c) 砜类和碳酸酯分子和溶剂化分子的氧化稳定性对比[6];(d) PP13TFSI离子液体电解液循环稳定性[43]Fig. 4 Schematic diagram of (a) Leakage currents during 4.5 and 4.6 V constant voltage floating tests of Li/LCO cells using different electrolytes [23], (b) the effect of TFP electrolyte in the cells[42]. (c) Comparison of oxidation stability between sulfone and carbonate molecules and solvated molecules [6]. (d) Cycle performance of ionic liquid electrolyte with PP13TFS[43] |
3.1.1.2 腈类溶剂
3.1.1.3 砜类溶剂
3.1.1.4 离子液体
3.1.2 新型锂盐
3.1.2.1 Al集流体腐蚀

图5 (a)LiPF6和LiFSI对Al集流体作用示意图;(b)3~4.6 V LiFSI电解液循环100圈后NCM811颗粒和Al集流体的SEM图[63];(c)传统电解液和LHCE对Al腐蚀的影响示意图[61]Fig. 5 (a) Schematic diagram of the effect of LiPF6 and LiFSI on Al current collector [65]; (b) SEM of NCM811 particles and Al collector after 100 cycles of 3~4.6V with LiFSI electrolyte [63]; (c) Schematic diagram of the influence of traditional electrolytes and LHCE on Al corrosion[61] |
3.1.2.2 局部高浓度电解液
3.1.2.3 多盐电解液
表1 新型电解液体系电化学性能Table 1 Electrochemical performance of novel electrolyte |
| Strategy | Electrolyte | Electrochemical system | Cut-off Voltage/V | Capacity Retention | Ref |
|---|---|---|---|---|---|
| Fluorinated | 2 mol/L LiPF6- DMC/FEC/FEMC | LiCoO2/Graphite | 4.5 | 74.2%(0.5 C,270 cycles) | 23 |
| 2 mol/L LiPF6- DMC/FEC/FEMC | LiNi0.5Mn1.5O4/Li | 5 | 89.9%(0.5 C,300 cycles) | 23 | |
| 1 mol/L LiPF6-FEC/FEMC/HFTFE | LiNi0.92Co0.04Mn0.04O2/Li | 5 | 68%(1 C, 500 cycles) | 24 | |
| 1.2 mol/L LiPF6-FEC/DFEC/FEMC | NCM811/Li | 4.4 | 90.8%(0.5 C, 200 cycles) | 25 | |
| 1 mol/L LiPF6-FEC/MTFP | NCM811/Li | 4.5 | 80%(0.5 C, 250 cycles) | 29 | |
| 1 mol/L LiTFSI- MDFA/MDFSA/TTE | NCM811/Graphite | 4.5 | 80.1%(0.5 C, 400 cycles) | 30 | |
| Nitrile | 1 mol/L LiDFOB-ADN/DMC | NCM111/graphite | 4.5 | 77%(0.5 C, 40 cycles) | 41 |
| 10 mol/L LiFSI-AN+VC | NCM111/Li | 4.5 | 85%(3.6 mAcm-2, 40 cycles) | 44 | |
| 1 mol/L LiTFSI-FEC/PN | NCM622/Li | 4.5 | 75.3%(1 C, 300 cycles) | 42 | |
| LiFSI-SN/AN | LiNi0.5Mn1.5O4/Li | 4.9 | 90.4%(0.1 C,100 cycles) | 46 | |
| NCM811/Li | 4.4 | 73%(0.1C, 200 cycles) | 46 | ||
| Sulfone | 1.2 mol/L LiPF6-FEC/TFPMS | NCM622/graphite | 4.5 | 71%(0.5 C, 400 cycles) | 49 |
| 1 mol/L LiPF6-EMS/DMC | LiNi0.5Mn1.5O4/Li | 4.9 | 97%(0.2 C, 100 cycles) | 50 | |
| 1 mol/L LiFSI-SL/FEC/HFE | NCM811/Li | 4.7 | 84.95%(0.5 C, 150 cycles) | 51 | |
| 1 mol/L LiTFSI-TMS/FEC | NCM811/Li | 4.4 | 86%(0.5 C, 500 cycles) | 48 | |
| Ionic-Liquid | 2.4 mol/L LiTFSI/PMP-FSI | LNMO/graphite | 5 | 85%(0.5 C, 200 cycles) | 56 |
| 1 mol/L LiFSI+0.3 mol/L LiNO3-PP13TFSI/DME | NCM811/Li | 4.3 | ~72%(5 C, 600 cycles) | 43 | |
| LHCE | LiFSI-DME/FEC/PFPN | NCM811/ graphite | 4.6 | 89.8%(0.33 C, 300 cycle) | 65 |
| 2.0 mol/L LiFSI/DME-BTFMD | NCM811/Li | 4.4 | 87%(1 C, 250 cycle) | 69 | |
| 1 mol/L LiFSI/TMS-TTE | LNMO/Li | 4.9 | ~93%(1 C, 100 cycles) | 70 | |
| Dual Salt | 1 mol/L LiPF6+0.1 mol/L LiFSI+0.1 mol/L LiBF4 | Li1.2Mn0.54Ni0.13Co0.13O2/Li | 4.8 | 80.4%(1 C, 400 cycle) | 73 |
| 0.8 mol/L LiFSI+0.1 mol/L LiTFSI+ 0.6 mol/L LiPF6-EMC | NCM811/graphite | 4.5 | 82.1%(1 C, 200 cycle) | 74 |
3.2 构建稳定阴极CEI层的进展
3.2.1 磷酸盐类添加剂
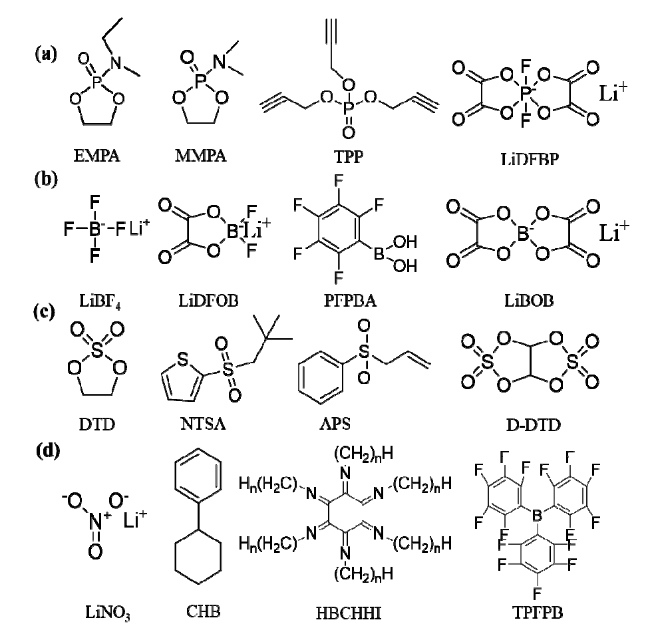

3.2.2 硼酸盐添加剂
3.2.3 含硫添加剂

3.2.4 其他添加剂
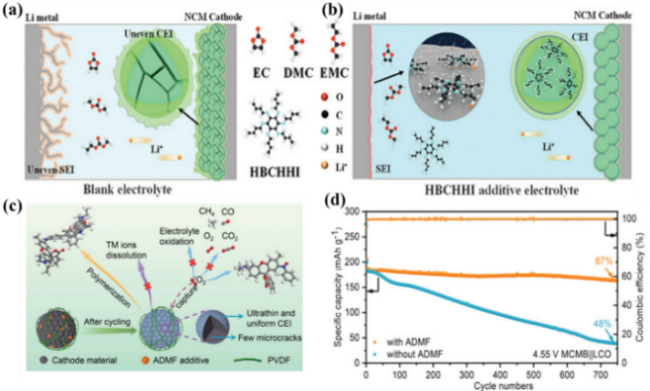
图9 (a)空白电解质和(b)含HBCHHI的电解质中SEI和CEI的保护机制示意图[102];(c)ADMF添加剂作用机理[103];(d)在0.5C、3.0~4.55 V含或不含ADMF的LCO/MCMB全电池的循环性能和库仑效率[103]Fig. 9 Schematic illustration of the protection mechanism of the SEI and CEI in (a) blank electrolyte and (b) HBCHHI-contained electrolyte [102]. (c) Schematic diagram of the mechanism of ADMF additive [103]. (d) Cycling performance and Coulombic efficiency versus cycle number of LCO/MCMB full-cells with or without ADMF at 0.5 C at 3.0~4.55 V [103]. |
表2 高压电解液成膜添加剂电化学性能Table 2 Electrochemical performance of typical additive in high-voltage electrolyte |
| Additive | Electrolyte | Electrochemical system | Cut-off Voltage/V | Capacity Retention | Ref |
|---|---|---|---|---|---|
| Phosphorous additive | 1 mol/L LiPF6-EC/DEC+0.5wt%EMPA | NCM523/Gr | 4.5 | 63% (0.5 C, 400 cycles) | 76 |
| NCM111/Gr | 4.5 | 200 cycles) | |||
| 4.6 | 300 cycles) | ||||
| 1 mol/L LiPF6-EC/DEC+1%TPP | NCM523/AG | 4.6 | 88.2%) | 77 | |
| 1.2 mol/L LiPF6-EC/EMC+1%TTFP | NCM523/graphite | 4.6 | 99.7% (C/3, 50 cycles) | 75 | |
| 1.3 mol/L LiPF6-EC/EMC/DMC+1%LiDFBP | Li1.17Ni0.17Mn0.5Co0.17O2/Li | 4.6 | 90% (0.5 C, 100 cycles) | 80 | |
| Boronated additive | 1 mol/L LiPF6-EC/EMC+0.1%TEAB | NCM811/Li | 4.3 | 63.2% (100 cycles) | 81 |
| 1.3 mol/L LiPF6-EC/EMC/DMC+1%LiDFOB | Li1.17Ni0.17Mn0.5Co0.17O2/graphite | 4.7 | 82.7% (0.5 C, 100 cycles) | 82 | |
| 1 mol/L LiPF6-EC/DMC+2%LiBOB | Li1.18Ni0.18Mn0.55Co0.09O2/graphite | 4.7 | 89.5% (0.2 C, 200 cycles) | 85 | |
| 1.0 mol/L LiPF6-EC/DMC+1%LiBOB | LiNi0.83Co0.11Mn0.05B0.01O2/Li | 4.6 | 73.1% (1 C, 200 cycles) | 88 | |
| LiDFOB+LiPO2F2+LiPF6-EMC/DMC | NCM811/Li | 4.7 | 80% (1 C, 500 cycles) | 89 | |
| 1 mol/L LiPF6-EC/EMC+5%PFPBA | NCM622/Li | 4.6 | 91.2% (1 C, 400 cycles) | 90 | |
| S-containing additive | 1 mol/L LiPF6-EC/EMC +1%PS | NCM811/Li | 4.45 | 80% (0.5 C, 200 cycle) | 93 |
| 1 mol/L LiPF6-EC/EMC +1%P-DTD | NCM811/graphite | 4.4 | >90% (1 C, 500 cycles) | 96 | |
| 1 mol/L LiPF6-EC/EMC +1%MMDS | NCM523/graphite | 4.5 | 92.78% (1 C, 800 cycle) | 97 | |
| 1 mol/L LiPF6-EC/EMC +1%MMDS | NCM523/graphite | 4.6 | 800 cycle) | ||
| 1 mol/L LiPF6-FEC/EMC/DMC +1% NTSA | LiCoO2/ω-LVO | 4.35 | 94.5% (200 cycle) | 99 | |
| 1 mol/L LiPF6-EC/EMC/DMC +FEC+EVS | Li1.17Ni0.265Co0.047Mn0.517O2/Li | 4.8 | 97% (1 C, 300 cycles) | 100 | |
| 1 mol/L LiPF6-EC/DMC +0.5%BFS | LiCoO2/Li | 4.6 | 88% (1 C, 300 cycles) | 101 | |
| Other additive | 1 mol/L LiPF6-EC/EMC/DEC +5m mol/L HBCHHI | NCM811/Li | 4.7 | 63.6% (5 C, 500 cycles) | 102 |
| 1 mol/L LiPF6-EC/DMC/DEC +1%SN+0.1%CHB | LiCoO2/Li | 4.6 | 77.4% (1 C, 500 cycles) | 104 | |
| NCM811/Li | 4.7 | 600 cycles) | |||
| 1 mol/L LiPF6-EC/DMC +1%ADMF | LiCoO2/MCMB | 4.55 | 87% (0.5 C, 750 cycles) | 103 | |
| NCM811/MCMB | 4.4 | 750 cycles) | |||
| 1 mol/L LiPF6-FEC/EMC +1%TPFPB+3wt%LiNO3 | LiCoO2/Li | 4.6 | 89.8% (0.2 C, 160 cycles) | 105 |
3.3 H2O和HF自清除的研究
3.3.1 Si—O/Si—N官能团清除H2O和HF
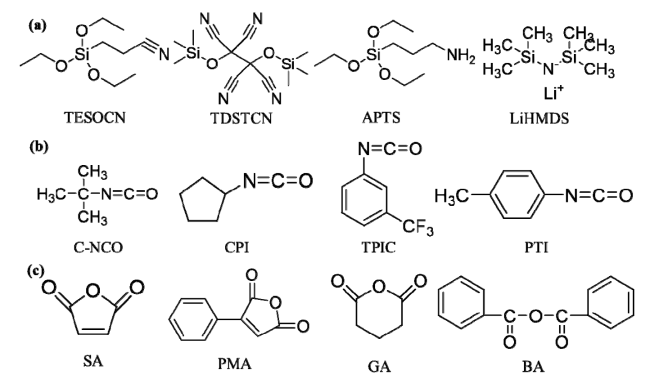
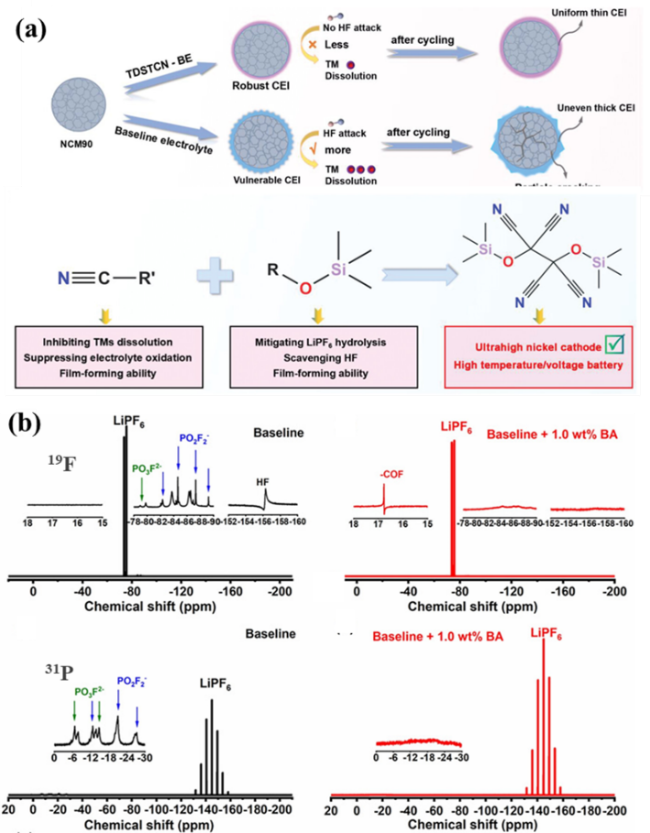
图10 (a)含有Si—O和Si—N官能团的化合物;(b)含有—NCO官能团的化合物;(c)含有酸酐官能团化合物的结构式Fig. 10 The molecular structures of (a) Compounds containing Si—O and Si—N functional groups. (b) Compounds containing —NCO functional groups. (c) Compounds containing acid anhydride functional groups |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}