




1 引言
氨(NH3)作为世界上最重要的化学产品之一,它可以生产氮肥,是高效的可再生燃料,还是一种无碳的能源储存中间体,由于这些优点及需求,NH3一直被大量生产。光催化固氮是在环境条件下光催化生产氨,因其绿色、环保而被广泛研究[1],它利用可持续能源作驱动力,水作绿氢原料,工艺零碳排放,是未来“绿氨”的发展方向。而且地球上可以轻松采集的太阳能(每年约5 × 1022 J)是完全可以满足全球能源需求的[2]。通过光催化剂,太阳能被直接收集并耦合到催化反应中进行化学转化,大大降低了能源需求[3,4]。自从有光催化N2还原的开创性报道以来[5],为了实现该技术的发展,科研人员研究探索了大量的光催化材料,然而氨生产效率仍然不令人满意,离工业应用还有很大的差距。因此,迫切需要开发新型材料以提高光催化固氮效率。
利用取之不尽的太阳能光催化固氮是一种绿色、有前景的人工固氮途径。光催化N2还原(NRR)的过程可分为以下步骤:(1)半导体催化剂对氮气进行吸附;(2)半导体接受光照产生光生电子,光生电子被激发后跃迁到导带,并在价带留下空穴,光生电子具有较强的还原能力,光生空穴具有较强的氧化能力,但大部分光生电子和空穴会发生无效复合;(3)同时,未复合的电子和空穴迁移到催化剂表面,参与表面的氧化还原反应。H2O被空穴氧化释放出质子,吸附在催化剂表面的N2结合光生电子和水质子后被还原成NH3。光催化固氮过程如图1所示。
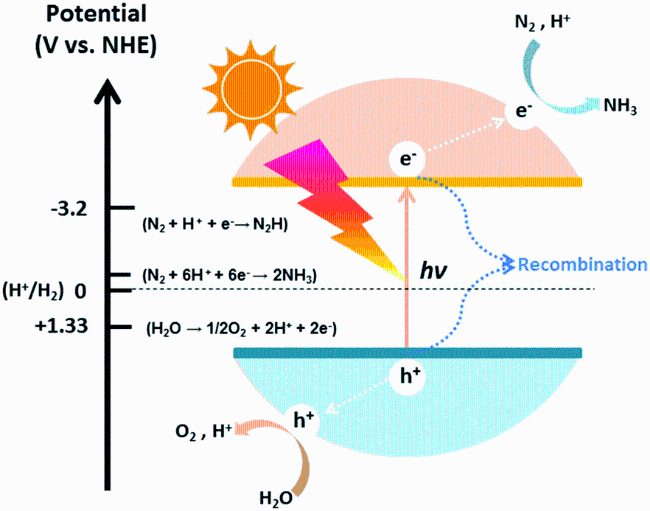
本文以提升光催化固氮活性为目的,通过以下3个方面对构建光催化剂策略进行归纳总结:(1)通过构建合适的催化剂活性位点,促成氮气的吸附和活化,从而保证后续反应的顺利进行;(2)通过强化电荷转移能力实现载流子的高效分离和应用,使其有效地参与表面反应,从而提升催化剂的氧化还原能力;(3)通过调节催化剂的表面状态提升催化剂对光的吸收与利用。
2 N2吸附和活化能力的提高
在光催化NRR体系中,氮分子在光催化剂活性位点上的有效扩散是后续N2化学吸附的前提条件,由于N≡N键解离能高达941 kJ·mol−1,N2的吸附和活化十分困难,因此,光催化固氮的主要挑战是催化剂与N2的结合和强N≡N键的激活[28,29]。因此选择合适的活性位点对N2吸附和活化至关重要,通过构建活性位点促进电子向N2转移是活化N2的一种有效策略[30]。活性位点可以定义为催化剂表面位点,它与反应物或反应中间体相互作用,通过影响其吸附、催化转化和解吸过程来提高催化性能。活性位点的选择和活性位点的配位/化学环境调节在光催化活性的增强中起着至关重要的作用,它们能够降低活化能势垒,从而促进反应的进行[31]。在光催化固氮过程中,首要前提是确保催化剂表面具有很好的氮气吸附能力,氮气被活化后加氢还原,参与还原反应的氮和氢越多,光催化固氮效率就会越高。目前NRR体系中氮气的吸附和活化位点有空位、Fe金属位、Ru位点等,因此围绕这些活性位点,科研工作者做了大量的工作。
2.1 引入缺陷位点
阴离子空位在氮气固定的光催化剂表面缺陷工程中占有重要地位,削弱N≡N的一种可行方法是制造富含电子的活性中心,而阴离子空位则是通过减少相邻配位非金属元素的数量,直接构建了富电子中心,当吸附氮气后,过渡金属(TM)通过σ键与N2结合,TM未占据的d轨道接受N的已占据p轨道的电子密度,然后将TM的已占据d轨道的价电子提供给N的π*,从而削弱N—N键,这种强π反馈机制是利于氮气的活化[32,33]。制造表面氧空位(OV或VO)或其他阴离子空位(C、N和S空位)是获得高效光催化固氮的有效策略之一。空位充当了富电子中心,通过在催化剂中引入不同的空位,如氧空位(OVs)、氮空位(NVs)和硫空位(SVs),可以有效地调节材料的电子结构、电荷输运和表面吸附能力,这些空位是多相催化的主要吸附和活性位点,能降低活化能垒,促进催化反应。
大量研究发现氧空位是重要的空位之一,对于NRR反应具有很好的效果,氧空位和氮气具有强吸附作用,有利于氮气的吸附;氧空位还是电子捕获位点,通过供电子促进氮气的还原[34⇓⇓⇓⇓~39]。Wang等[18]设计了含有不对称氧空位缺陷的MoO3−x纳米线,OVs引入的缺陷诱导了电荷的重新分配,从而增强了N2的吸附和活化。同时,在光催化固氮过程中,不对称缺陷的结构通过纳米尺度上的电荷重新分配促进了光生电子-空穴对的分离。Li等[40]制备的锌掺杂Co3O4多面体,表面有丰富的氧空位,这些氧空位不仅为氮的吸附和活化提供了活性位点,而且增强了光载体的分离能力,显著提高了材料的光催化固氮效率。Sun等[41]构建了VO-BiOBr/TiO2,并全面研究了氧空位对VO-BiOBr/TiO2的光吸收、载流子寿命、电荷转移、光电化学性能和光电催化性能方面的影响,研究发现配位不饱和态的氧空位导致BiOBr禁带出现缺陷水平,导致光吸收增强,同时氧空位作为光生电子的捕获中心,激活吸附在催化剂表面的N2,TiO2与VO-BiOBr之间的异质结降低了界面转移阻力,有利于光生载流子转移,从而提高了光电催化NRR活性。
硫空位是常见的一种阴离子空位,它能极大地促进光响应特性[17,42,43]。硫空位在半导体催化剂中起到主反应中心的作用,同时利于分离电子-空穴对和加速载流子迁移。Tian等[44]制备的SV-1T-MoS2光催化剂,其优势在于存在较多的硫空位和1T相,具有较多的活性边位和良好的金属导电性,具有较高的氮吸附和活化硫空位含量,增强了光吸收强度和吸收区域,具有较好的电荷分离和转移能力,有利于光催化固氮。材料中硫空位的含量对N2的光催化转化也有很大的影响,为了探索其影响,Lan等[45]通过改变溶剂热反应时间来控制Bi2S3纳米棒中硫空位的含量,当反应时间为3 h时,Bi2S3中硫空位浓度达到最优值。此时,Bi2S3-3纳米棒的N2捕获能力、光响应范围和氮转化效率均达到最佳水平。
表1 缺陷材料的光催化固氮性能Table 1 Photocatalytic nitrogen fixation performance of defective materials |
| Photocatalyst | Reaction medium | Light source | NH3 evolved | Defect type | Year | Ref. |
|---|---|---|---|---|---|---|
| a-MoO3-x NWs | Water | 300 W xenon lamp | 200.35 μmol·h-1·g-1 | OVs | 2023 | 18 |
| Zn-Co3O4 | Water | 300 W xenon lamp (λ ≥ 420 nm) | 96.8 μmol·h-1·g-1 | OVs | 2023 | 40 |
| SV-1T-MoS2 | Water+ Methanol | 300 W xenon lamp (AM 1.5G) | 8220.83 μmol·h-1·g-1 | SVs | 2020 | 44 |
| Bi2S3 | Water | 300 W xenon lamp | 51.04 μmol·L-1·h-1 | SVs | 2022 | 45 |
| NYF/NV-CNNTs | Water + Sacrificial agent | 300 W xenon lamp (λ ≥ 420 nm) | 1.72 mmol·L-1·g-1 | NVs | 2021 | 48 |
2.2 引入金属位点
(1)金属Fe
有研究表明,通过在半导体中引入过渡金属能促进N≡N的裂解过程,可以获得较高的光催化氮还原活性和氨合成能力[51⇓⇓~54],其中Fe是公认的理想的N2还原位点。无论生物固氮还是人工固氮,铁都起关键作用。对生物固氮体系的研究表明,FeMo蛋白酶中,Fe是N2结合和还原的优先活性位点[55]。在工业固氮过程,铁也是主要的催化剂,关键步骤涉及N2在Fe表面的化学吸附和Fe—N络合物的形成。因此过渡金属Fe被认为是工业和生物固氮中重要的催化活性位点[56]。在光催化固氮体系中,Fe具有多重优势。首先,Fe不仅影响催化剂的晶体结构,还有助于捕获光生电子和空穴,并抑制它们的复合,这种电子-空穴分离过程可以有效地提高光催化固氮的效率[57]。其次,由于N2分子具有孤对电子,而Fe的空d轨道可以接收这些孤对电子,此外,Fe原子拥有独立的d电子,可以贡献到反键轨道上,从而增强Fe—N键,破坏N≡N键。这种Fe—N键的增强对于有效捕获和活化N2非常重要,因此铁基材料具有较高的光催化固氮活性,在光催化固氮领域有大量的研究。
Azofra等[58]的理论计算结果表明,在Fe-MoS2体系中,通过引入Fe中心,与N2分子形成更强的相互作用,可以显著提高N2分子在催化剂上的化学吸附能力,这种增强的吸附能力有助于降低N2的活化能势垒,促进电子从催化剂向N2分子的转移,进而实现N2的活化而促进固氮性能的提高。Shen等[57]制备了FeⅢ掺杂的花状BiOCl,其活性位点曝光高,具有良好的N2光固定性能。FeⅢ不仅是N2活化的反应中心,也是BiOCl中捕获和迁移电子到N2分子的“电子转移桥”。Zhao等[59]制备了一种Fe掺杂改性的TiO2催化剂,并研究了其在光催化固氮中的性能。Fe掺杂可以加速电子和空穴的转移,防止复合,并促进光催化固氮过程中N2的吸附和活化,催化剂的光催化活性得到提高。Liu等[60]制备了Fe掺杂的BiOBr光催化剂,以BiOBr为基质,引入Fe活性位点,来提高固氮效率。Fe既是N2吸附和活化的活性位点,同时Fe掺杂促进晶体形成了更多的OVs,并且根据理论计算发现,OVs连接的Fe原子会从附近的其他原子中抽走光激发电子,形成富电子的Fe(Ⅱ)。Fe(Ⅱ) 3d轨道上多余的电子将通过电子反馈注入被吸附N2的π N—N反键轨道,最终使固氮效率提高了8倍。Zhao等[61]制备了Fe掺杂的SrWO4纳米颗粒,发现Fe掺杂使SrWO4的本征带隙大大缩小,不仅扩大了从紫外到可见光的吸收范围,而且减少了电荷复合,Fe掺杂还将FeⅡ/FeⅢ氧化还原途径引入到NRR的表面中心,最终提高NRR效率。部分Fe掺杂文献的固氮条件及产率总结见表2。
表2 Fe掺杂材料的光催化固氮性能Table 2 Iron-doped materials for photocatalytic N2 fixation |
| Photocatalyst | Reaction medium | Light source | NH3 evolved | Year | Ref. |
|---|---|---|---|---|---|
| Fe-BiOCl | Water | 300 W xenon lamp | 30.00 μmol·L-1·h-1 | 2020 | 57 |
| Fe-BiOBr | Water | 300 W xenon lamp (λ≥ 420 nm) | 382.68 μmol-1·g-1·h-1 | 2020 | 60 |
| Fe-SrWO4 | Water | 300 W xenon lamp | 150.70 μmol·h-1·g-1 | 2020 | 61 |
| Fe0.05-CN | Water + Alcohol | 250 W high voltage sodium lamp | 5.40 mg·L-1·h-1·g-1 | 2017 | 62 |
| Fe-MCNC | Water + Methanol | 65 W LED | 1.88 μmol·h-1·g-1 | 2023 | 63 |
(2)金属Ru
Ru被认为是第二代NH3合成催化剂,由于Ru的N2还原电位低于Fe[64],Ru原子能将电子从其d轨道转移到反键轨道上,促进N2的解离化学吸附,从而加速反应进行。与Fe相比,Ru更有利于N≡N的活化,因为它需要的反应条件相对温和[65,66],这和光催化环境是相一致的,因此认为Ru是比Fe更好的光催化NH3合成催化活性位点[67,68]。首先,负载Ru的活性位点可以显著提高光催化活性。这是因为Ru与含氧载体之间的相互作用可以构建Ru-O-M (M指其他金属)位点[69⇓~71]。同时,这样的相互作用能够促进电子向被吸附的N2转移,进一步提高光催化固氮效率。其次,Ru纳米粒子在光激发下能够产生光生电子,并能够改变其他金属周围的电子结构[72,73],这使得Ru广泛应用于光催化固氮反应中。在光催化固氮领域,已经有很多报道使用Ru负载对催化剂改性来提高光催化活性并提高反应效率。
Liu等[74]将Ru单原子分布在非晶态氧化铁纳米片(Ru1/2DAF)上,非晶化的氧化铁能够调节电子态密度,降低电子的转移能垒,通过d(Ru)-d(Fe)的耦合构建电子通道,将电子从非晶态载体引导到Ru 4d轨道上,加速光生电子在Ru活性位点上的富集,从而促进N2的吸附和活化,最终提高反应活性,图2a为Ru1/2DAF的光生电子示意图。Ding等[75]构建了Ru-MOF-74体系,研究发现Ru没有取代MOF-74中的Zn,而是存在于内部通道中,引入Ru促进了N2的吸附,延长了可见光响应,促进了载流子的分离,使载流子有效的转移到催化剂表面,从而提高了光催化固氮性能。Li等[76]设计了Ru团簇辅助Ⅲ-氮化物纳米线Ru@GaN用于光催化固氮,金属Ru与基底形成界面肖特基,使电子从Ⅲ-氮化物转移到Ru上,Ru充当了电子池,降低了N2解离反应的活化势垒,其形成的肖特基势垒的示意图如图2b所示。Liu等[77]设计了单原子Ru修饰的富氧空位TiO2纳米片,实验结果证实了单一Ru位点可促进N2光还原生成NH3,Ru原子削弱了析氢,促进了N2的吸收,并改善了载流子分离,从而增强了N2光固定,其N2光还原机理如图2c所示。Li等[30]设计的Ru/W18O49,构建了不对称的活性位点,Ru-O-W中心可作为N2吸附位点,能更好地促进电子向被吸附的N2转移,而Ru与氧空位间的等离子共振效应为氮气活化提供足够的电子,两者共同作用达到了提高光催化固氮活性的目的。以上文献的固氮条件及产率总结见表3。

图2 (a) Ru1/2DAF的光生电子转移示意图[74]; (b) n型GaN NWs与金属Ru团簇形成肖特基势垒的示意图[76]; (c) 含氧空位的Ru负载TiO2纳米片上N2光还原机理图[77]Fig. 2 (a) Schematic diagram of photogenerated electron transfer in Ru1/2DAF[74]. (b) Schematic diagram for the formation of the Schottky barrier between n-type GaN NWs and metallic Ru clusters[76]. (c) Mechanistic diagram of N2 photoreduction over single Ru site loaded TiO2 nanosheets with oxygen vacancies[77] |
表3 Ru负载材料的光催化固氮性能Table 3 Photocatalytic nitrogen fixation of Ru supported materials |
| Photocatalyst | Reaction medium | Light source | NH3 evolved | Year | Ref. |
|---|---|---|---|---|---|
| Ru/W18O49 | Water | 300 W xenon lamp | 44.30 μmol·g-1·h-1 | 2023 | 30 |
| Ru1/2DAF | Water | 300 W xenon lamp | 213.00 μmol·g-1·h-1 | 2022 | 74 |
| Ru-MOF-74 | Water | 300 W xenon lamp (λ ≥ 420 nm) | 70.90 μmol·g-1·h-1 | 2022 | 75 |
| Ru@GaN | Hydrogen + Nitrogen | UV lamp 290-380 nm | 2400.00 μmol·g-1·h-1 | 2017 | 76 |
| Ru-TiO2 | Water + Alcohol | 300 W xenon lamp | 56.30 μg·h-1·g-1 | 2019 | 77 |
3 电荷转移能力的提升
催化剂改性的另一目标是提高电荷的转移能力以促进电子-空穴的有效分离,进而加速氧化还原反应的进行,最终提高光催化固氮的效率。构建具有电荷转移能力的催化剂可以通过以下方式完成。
3.1 构建单金属混合价态催化剂
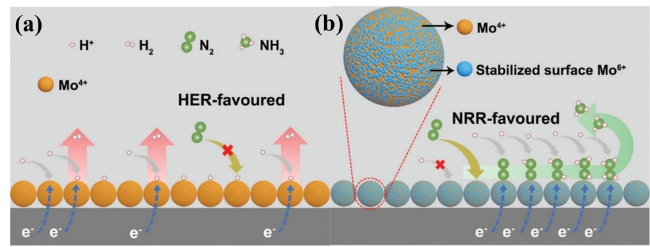
利用低价钴的形成产生氧空位(OVs),通过构建Co2+/Co3+循环提高催化活性。而且Co2+/Co3+循环通过光激发产生,光照结束后能够恢复如初。在光催化固氮反应中,金属活性中心通过吸收光能进行氮气的吸附和活化后进行一系列氧化还原反应,并转化为可利用的氨。在光照条件下,金属活性中心的氧化还原循环能在不同的氧化态和还原态之间进行转换。因此,加速金属活性中心的变价循环能提高光催化固氮的氧化还原能力,而构建具有高还原态/氧化态比例的催化剂则是提高氧化还原反应的前提条件。
铁金属是一个具有典型混合价态的金属,在生物固氮的固氮酶中,FeMo因子的铁是以FeⅡ和FeⅢ混合价态形式存在的[83],FeⅡ是氮气的吸附和活化位点,FeⅢ不参与氮气还原过程,但是保证了酶团簇结构的稳定性[84]。在工业合成氨中Fe1-xO基催化剂的高活性是由于FeⅡ和FeⅢ共存[85]。不论生物固氮还是工业固氮,都是FeⅡ和FeⅢ混合价态共存体系,并且其价态可以在FeⅢ和FeⅡ之间进行可逆转换。由于FeⅡ与不饱和分子具有更强烈的相互作用[86],它作为氮气的吸附和活化位点,提高FeⅡ比例有利于固氮反应的进行。Jiang等[87]设计了MIL-53(FeⅡ/FeⅢ)光催化剂用于光催化固氮研究,通过一步溶剂热法,将MIL-53(FeⅡ/FeⅢ)中的FeⅢ部分被乙二醇(EG)原位还原为FeⅡ,构建了具有FeⅡ和FeⅢ混合价金属簇的光催化剂,并且通过实验证实了FeⅡ/FeⅢ比是影响MIL-53(FeⅡ/FeⅢ)催化活性和骨架稳定性的关键参数。
3.2 设计双金属位点催化剂
受生物氮化酶的“FeMo辅因子”的启发,Li等[91]构建了一种新型仿生Fe/Mo双金属光催化剂,它能够通过多电子氧化还原反应有效地促进光生载流子的分离和运输,使载流子寿命延长2.8倍,固氮活性提高4.8倍。Ni等[92]构建了CeF3:Yb3+, Er3+/Fe-ATP异质结构的光催化剂进行全谱固氮,CeF3中的氟空位(FV)与ATP中的Fe形成双活性位点,促进氮分子的活化,有效降低光催化固氮反应中惰性N2的活化能。活性氮转移到Fe-ATP表面,形成Fe—N=N键,使其更容易加氢,在光生电子参与后,Fe—N=N成为新的反应中心,而FV作为电荷介质,建立了间接的Z型结构,促进了电荷的分离,最终得到较高的固氮活性。Scholten等[93]构建了双金属RuPd纳米颗粒支撑在石墨氮化碳上的可持续催化系统用于光催化固氮,Ru削弱N≡N键,Pd键合H,两种金属间有利的协同作用,导致其具有独特的催化性能。固氮原理如图5所示。表4列出了部分双金属位点材料在光催化固氮领域的应用。
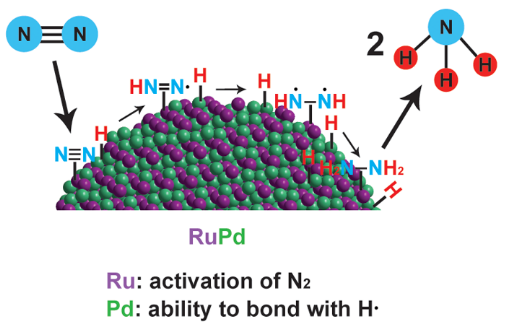
表4 双金属位点材料的光催化固氮性能Table 4 Bimetallic site materials for photocatalytic N2 fixation |
| Photocatalyst | Reaction medium | Light source | NH3 evolved | Year | Ref. |
|---|---|---|---|---|---|
| Fe/Mo-BMWO | Water | 300 W xenon lamp (λ ≥ 420 nm) | 218.93 μmol·h-1·g-1 | 2022 | 91 |
| CeF3:Yb3+, Er3+/Fe-ATP | Water | 300 W xenon lamp | 253.60 μmol·h-1·g-1 | 2022 | 92 |
| RuPd/C3N4 | Water + Alcohol | 300 W xenon lamp | 1389.84 μmol·h-1·g-1 | 2021 | 93 |
| Ru-SA/ HxMoO3−y | Hydrogen + Nitrogen | 300 W xenon lamp (λ≥ 420 nm) | 4.00 mmol·h-1·g-1 | 2022 | 94 |
| RuO+Co/CoO | Water | 300 W xenon lamp (λ≥ 400 nm) | 306.00 μmol·h-1·g-1 | 2022 | 95 |
受以上研究的启发,结合光催化固氮的加氢机制,构建具有不同功能作用的双金属体系是一种可行的思路。利用双金属间的电荷流动来加速氧化还原反应,不同金属在固氮过程中分别完成氮气活化和加氢过程,从而协同配合完成整个反应过程,进一步提高催化活性。
3.3 构建多金属位点催化剂
与传统的单金属光催化剂相比,多金属位点光催化剂在催化性能和反应机理上具有独特的优势和应用潜力。它能够利用不同金属各自的特性,如不同的电子结构、能带结构和配位特性等,实现更丰富、多样的反应路径和反应机制。这种多金属协同作用可以提高催化剂的电荷转移能力、氧化还原能力和催化活性,从而增强光催化反应的效率和选择性。Ma等[96]通过调节光激发电子的转移路径,改善了(Ru/WC)/CdS催化剂上的光催化析氢性能。Ru能快速转移电子,是生成氢的催化活性中心,WC具有较大的比电容,CdS是收集和存储光生电子的,明确了光激发的电子转移路径是依次从CdS到WC和Ru上。Zhang团队[97]构筑了K/Ru/TiO2-xHx多金属位点固氮催化剂实现了高效太阳能合成氨,K和富含氧空位(OVs)的富电子载体TiO2-xHx都可以调节Ru的电子结构,使其有效地活化氮。此外,由于TiO2-xHx的界面结构接受来自Ru的氢原子,使得催化剂表面具有更好的活性。在氮气固定的同时,该界面还能将氢气中毒的问题降至最低。界面上的TiO2-xHx接受来自Ru的H原子,并将其传递给Ru活化的N2上,最终形成Ti-NHx。
4 提高光利用率
Zhu等[105]设计了核壳结构的SiO2@ZnS/CuSx催化剂,入射光通过ZnS/CuSx壳层后,被白色SiO2芯层反射和散射,然后被ZnS/CuSx收集。图6a的核壳SiO2@ZnS/CuSx的光散射原理图。Zaine等[106]通过优化SiO2-TiO2核壳结构提高了光电极染料敏化太阳能电池的光散射效率。Bai等[107]设计的C/CdS@ZnIn2S4核壳结构的光催化剂用于CO2还原,将其优异的光催化性能归功于空心碳,首先多次的光反射和散射改善了光捕获,促进了电荷分离,利于电子收集,其次大表面积提供了更多的靶向还原反应的高活性和选择性位点,多孔壳在空间上分离还原和氧化半反应,同时保护了CdS免遭光腐蚀,核壳结构将光响应范围扩展到更长的波长,并通过电子和空穴在不同组分上达到空间分离的效果。图6b为C/CdS@ZnIn2S4核壳结构光催化剂的光利用示意图。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
光催化固氮以太阳能作驱动力,常温常压下利用N2和H2O直接产生NH3,工艺零碳排放,是极具前景的人工固氮方式之一,并且光催化固氮已取得一定进展,但性能与实际工业生产需求相距很大,因此提高其效率一直是研究者们努力的方向。本文主要从光催化剂改性的角度,结合光催化固氮的反应过程,较为详细地讨论并总结了提高光催化固氮效率的策略。为进一步提高光催化固氮效率,光催化剂的设计思路可以从以下方面进行深入研究。(1)N2的吸附和活化能力:N2在催化剂上的吸附和活化是固氮的第一步,构建具有高吸附、活化N2能力的活性位点是提高固氮效率的有效策略。结合先进的表征技术及理论研究方法,优选更精确和更有效率的活性位点,最大程度地活化N2,从而提高N2还原的效率。(2)电荷转移能力:提高光生电子的迁移能力,使其定向并快速地迁移至催化剂表面,高效地将N2还原。构建多种原位表征技术,实时监测并精准掌握光生电子迁移路径,提高光生电子的利用率。(3)太阳光利用率:可采用不同的提升策略来提高太阳光的利用率,包括设计催化剂表面结构、调控表面形貌等。
除了从光催化剂上着手提高效率外,可以在整个光催化固氮过程中进行改进,设计一个系统和标准化的光催化固氮系统来模拟工业生产过程,全面对固氮效率进行评价并优化。