




1 过氧化氢的工业生产方法
病房、救护车、机舱等环境消毒常用雾化H2O2或H2O2蒸气两种方式。雾化系统将浓度5%~6%的H2O2溶液加压,通过喷嘴喷出,产生0.5~10 μm直径的H2O2气溶胶,常用的剂量为6 mL/m3,消毒时间以2 h为宜[4]。已经有多种H2O2气化喷雾消毒机在市场上销售[1,5]。H2O2蒸气体系包括一个蒸汽发生器和H2O2浓度监控器,在消毒前通过除湿机降低空间的湿度,然后采用高温蒸发浓度大于30%的H2O2溶液产生H2O2蒸气,用高速空气流带动H2O2蒸汽在封闭的房间内循环[4]。H2O2喷雾也可用于口腔科消毒。Lazzarino课题组建议采用“0.28 mL1.5% H2O2鼻腔喷雾剂,每天两次,同时用3% H2O2溶液漱口1 min,每天两次”的方式降低呼吸道传染病的风险[6]。食品消毒的重要目标之一是分解黄曲霉 毒素,H2O2用于食品消毒一般需结合高温、紫外线、碱处理等方法,例如将6%的H2O2水溶液加热到80 ℃,浸泡脱脂花生粉30 min,可去除97%的黄曲霉毒素[7]。向地表水中投加H2O2,当其浓度达到0.3%后可将噬菌体杀灭6.5个log,而氯消毒只能完成3.0个log[8]。
2-乙基蒽醌法是当前规模化集中生产H2O2的主要方法,占全球和国内H2O2产量的95%和99%[9]。蒽醌法以氢气和氧气为原料,以2-乙基蒽醌和金属钯为催化剂,以重芳烃(1,2,4-三甲苯)为溶剂生产H2O2。在一定温度、压力下,2-乙基蒽醌溶解于重芳烃,然后在钯催化剂作用下和氢气反应生成2-乙基氢蒽醌,2-乙基蒽醌溶解度不高,转换成2-乙基氢蒽醌后溶解度增加。2-乙基氢蒽醌在一定温度、压力下与氧气反应生成2-乙基蒽醌和H2O2[10]。经萃取从混合液中提取H2O2水溶液,最后经过重芳烃净化得到合格的H2O2水溶液。蒽醌法直接产生H2O2溶液的浓度通常为27.5%[11],更高浓度(如35%、50%、70%)的H2O2溶液可通过蒸馏浓缩获得。
规模化制备H2O2的方法还有氢、氧直接合成法[14],这些方法不但要求使用H2、金属催化剂等危险或昂贵的原料,而且存在储存和运输的问题。
2 以氧气为原料的原位产H2O2方法
氧气吸附在电极表面的方式有两种,Bridge连接的氧气倾向于得四个电子产生水;Pauling连接的氧气得一个电子产生OOH基团,被强吸附的OOH基团再得三个电子产生水,被中度吸附的OOH基团能够得一个电子产生H2O2,因此,电极表面对OOH基团的吸附能是决定H2O2选择性的关键[17]。实现这一目标的关键是研制2e ORR选择性高的阴极材料,如掺杂碳电极[18]。此外,空气氛围室温常压下,水中饱和溶解氧浓度只有8.25 mg/L,理论上只能转化成浓度8.76 mg/L的H2O2水溶液,重量比为0.000876%,比消毒需求浓度低3个数量级以上,因此采用中空电极内曝气[19]、纯氧曝气[20]、增压供氧[21]、气体扩散电极[22]等方式增加氧气供应。目前文献报道的碱性溶液中产H2O2浓度已经超过了5%[23],达到了实际消毒的浓度要求。
提高H2O2产量必须考虑抑制副反应。电化 学氧还原产H2O2过程中存在包括阴极分解水产氢、4-电子氧还原产水、1-电子H2O2还原产·OH等多个竞争反应。化学氧还原发生在阴极,分解水副反应的产物之一是OH-,导致局部pH上升,碱性条件容易引发H2O2分解产生O2和H2O[27]。此外,阴极产生的H2O2扩散到阳极可被迅速被氧化产生O2和H+。因此进一步提高H2O2浓度必须有效抑制副反应和避免H2O2分解。常见的思路是提高阴极分解水产氢过电势,如上一段提到的通过杂原子掺杂抑制4e ORR和H2O2的还原分解,使用隔膜阻止产生的H2O2扩散到阳极,采用连续流形式避免OH-积累。
3 以水为原料的原位产H2O2的方法
3.1 水氧化产H2O2的电化学和光催化方法
电化学产H2O2的另一个途径是阳极氧化水,反应式如下:2H2O → H2O2 + 2H+ + 2e‾。水氧化是固液两相界面反应,仅以水为原料不需要供应氧气,相对于要求气液固三相界面的氧还原反应具有传质优势。提高水氧化产H2O2浓度的关键是抑制竞争反应。2电子氧化水(2e WOR)产H2O2的竞争反应主要是4电子氧化水(4e WOR产氧气,为了提高H2O2选择性必须抑制4电子产氧反应,主要方法是使用产氧过电势高的半导体电极,如BiVO4、TiO2、WO3、SnO2、CaSnO3、ZnO等[1,31],市售碳毡、碳布、碳纸[32]以及硼掺杂金刚石做阳极[33]也具有较高的产氧过电势,常用于氧化水产H2O2的研究。
另一种抑制4电子氧化反应的方法是调控中间体的吸附能。当*OH的吉布斯吸附自由能值(ΔG*OH)在1.6 ~ 2.4 eV之间时,H2O2为主导产物,理想情况下,1.76 eV为最优热力学值[34]。ΔG*OH值低于1.6 eV的材料的强吸附会导致O2析出,ΔG*OH值高于2.4 eV的材料的弱吸附会导致·OH的产生[34]。因此,合理调控OH的吸附是实现高效H2O2生产的可行途径。在足够正的电位下,4e水氧化生成的氧气泡脱离电极向上运动后破裂消失,如果这些氧气被限制在活性位点附近,那么积累的O2分子可以进一步与催化剂表面OH相互作用,从而能够调整结合强度向有利于2电子氧化方向发展[34]。Wang课题组[35]通过在催化剂表面涂上疏水聚合物来捕获靠近活性位点的原位产生的O2气体,观察到一旦生成的O2气体被限制在表面,其H2O2选择性显著增加。Sun课题组[36]则通过向弱吸附OH的TiO2晶格中掺杂特定数量的强吸附OH的钌原子来调控吸附强度,最优条件下10 min累积了20 mL浓度为422 mg/L的H2O2。
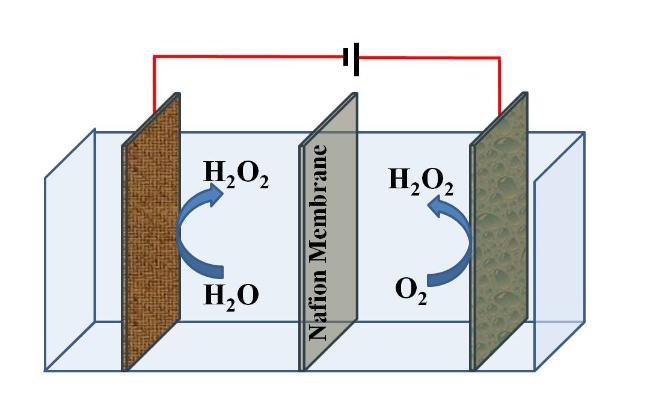
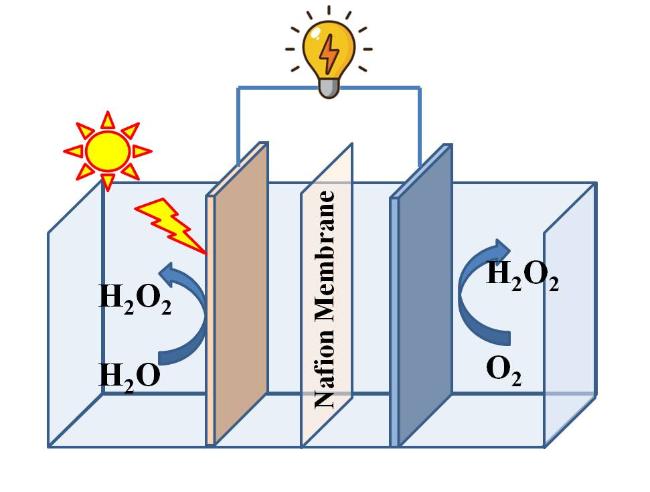
除此之外,还有一些以水为原料的H2O2现场制备方法,例如热催化法、超声压电催化法、等离子体裂解法、生物电化学法、微液滴法等,这些方法虽然报道不多,但提供了新思路或具有应用潜力,很有希望获得满足现场消毒要求的H2O2水溶液。本文总结了近年的最新报道,依次介绍这些方法。
3.2 热催化产H2O2
H2和O2直接合成产H2O2是众所周知的热催化反应[31,45]。由于H2和O2混合存在爆炸风险,需要找到安全的原料。水是理想氢源,与O2的直接氧化(H2O + 0.5O2 = H2O2 ΔG = 116.7 kJ/mol)是热力学上坡反应,因此需要输入能量[46]。常见的供能方式是热偶联,即用一个放热反应中释放的热量驱动一个吸热反应进行,例如用一氧化碳氧化成二氧化碳放出的热为金催化水和氧气反应生成H2O2反应供热[47]。液相热偶联的产物是H2O2与有机物的混合物,必须进行液相分离,分离过程的成本较高是这种方法的缺点。此外,这种方法大部分能量都在加热大量的稀释剂而不是用来推进所需的反应,对稀溶液来说能量效率很低。Kung等[48]提出在蒸汽相中进行热偶联反应产H2O2,这样可以提高反应物浓度,大幅提高能量效率。他们的研究表明,烯烃的环氧化和烷烃选择性氧化为醇等反应不能提供足够的热力学驱动力,而醇氧化为醛和酸释放的热量则可以驱动氧化水产H2O2反应。这种做法需要考虑高温条件下产生H2O2的自发分解。
热催化的另一种形式是热电材料利用温差引起的正负电荷分离将热能转化为电能产H2O2。热电电压是影响热电材料产H2O2浓度和体积的关键。在室温下,碲化铋(Bi2Te3)具有优异的物理性质和独特的电子结构,相对其他常见的热电材料如碲化锑(Sb2Te3)和碲化铅(PbTe)可产生更高的热电电压。Lin等[49]将碲化铋纳米板涂覆在碳纤维织物上制备出Bi2Te3@CFF材料。无论温差为正或负,Bi2Te3@CFF均表现出显著的抗菌活性与良好的耐久性。控制实验证实了在热催化反应过程中产生了有利于H2O2生成的超氧自由基,还证明了消毒能力来自原位产生的H2O2。具有高表面积二维结构是碲化铋纳米板(NPs)拥有优越热催化性能的原因。将基于Bi2Te3 NPs的抗菌过滤器安装在空调中,利用空调内部与外部环境的温差触发热催化反应。实验结果表明,温差为30 K时,5 mg碲化铋NPs可在20 min内产生30 μM H2O2。用吹风机产生温差,使用1× 1 cm Bi2Te3@CFF在20 min内针对1 mL浓度为2×106 CFU/mL的大肠杆菌的杀菌率达到72%,且稳定工作至少30 d[49]。
3.3 压电催化法产H2O2
钙钛矿是主要的压电材料,为增强压电极化,制造缺陷、掺杂金属元素和构建复合材料是有效途径。钙钛矿是一类分子通式为ABO3的氧化物。A和B分别为稀土(碱土)元素和过渡金属元素,A和B可被半径相近的其他金属取代而钙钛矿晶体结构不变。一般情况下,钙钛矿晶体中每个晶胞的净电荷均为零,但当晶胞中的钛离子略微偏离中心时,就会产生电极性,使晶胞转化为电偶极子。当机械应力作用在上时,钛离子的位置发生变化,进而改变晶体的极化强度,产生压电效应。
在产H2O2领域研究最广泛的钙钛矿是锆钛酸铅,分子式为Pb(Tix、Zr1-x)O3,由PbTiO3和PbZrO3固溶形成。通过改变外力调整锆钛酸铅的极化程度,可以调节局部电场的强度,从而调节锆钛酸铅表面对OOH基团的吸附能力,达到提高H2O2的产量的目的[52]。
为避免使用有毒的铅,研究人员探索了钛酸钡等钙钛矿压电催化材料的产H2O2性能。为了提高压电性能,三元钙钛矿锆钛酸钡也被开发出来,由于三相(正交、正方和菱形)共存,极化能垒降低,导致压电性显著增强,再加上适合的氧空位浓度,超声辐照下,锆钛酸钡的H2O2选择性达到80%。Li课题组[53]的研究进一步揭示了三元钙钛矿中压电极化促进电子-空穴对的产生、分离和输运,从而促进了催化反应。
压电催化性能取决于压电极化的强度,制造缺陷、掺杂金属元素和构建复合材料是强化压电极化的有效方法。Huang课题组[54]制备出厚度只有4.2 nm的超薄Bi4Ti3O12纳米片,这个厚度仅相当于1~2个Bi4Ti3O12晶胞,由于表面大量暴露,具有丰富的表面氧缺陷(OVs)。相对于普通Bi4Ti3O12,这种富含OVs的超薄Bi4Ti3O12纳米片对外力更加敏感,可产生更强的压电极化场,促进压电自由电荷的分离和迁移,延长了压电自由电荷的寿命,并降低了O2分子的吸附能,促进其活化为超氧自由基。得益于这些优点,具有最佳OVs浓度的超薄Bi4Ti3O12纳米片显示出优异的压电催化产H2O2生成性能,0.05 g催化剂加入到100 mL乙醇-水溶液中,在超声作用下2 h内可产生161 μmol的H2O2,换算成重量百分比为0.0054%,明显高于厚度25.8 nm的普通Bi4Ti3O12纳米片的86 μmol(0.0029%)[54]。
除了构建缺陷,对压电材料掺杂金属元素可 提高极化强度。例如将Nb5+掺杂进BaTiO3的晶格,可以引入电荷补偿,提高BaTiO3的载流子浓度和电荷迁移速率,从而在相同机械力作用下获得更高的极化电场强度。将压电效应与光催化作用耦合可以有效提升产H2O2能力,但钙钛矿材料一般具有紫外光响应的光催化能力,无法利用太阳光谱中比例最大的可见光,而负载碳量子点(CDs)通常可以帮助钙钛矿获得可见光响应能力。Zhai课题组[55]用金属铌掺杂钛酸钡(BaTiO3:Nb)强化压电极化,再负载CDs作为可见光敏化剂和电子受体提升产H2O2浓度。在可见光和超声共辐照下,15 mg CDs修饰Nb掺杂BaTiO3催化剂加入到30 mL乙醇-水混合溶液中生成H2O2的速率为1360 μmol/(g催化剂·h) (0.0023%),相对于BaTiO3的48 μmol/(g催化剂·h)提升了27倍[55]。
第三种强化极化电场的方法是构建复合材料。一种用球形硫化锌和纳米压电钛酸钡修饰的多层In2S3纳米片(ZnS/In2S3/BS3/BaTiO3)构建的复合材料在可见光和超声(压光催化)共辐照下,100 min内产生约378 µM的H2O2(0.0013%),连续反应600 min,ZnS/In2S3/BaTiO3产生的H2O2浓度可达1160 µM(0.0039%)。在压光催化下,H2O2产率的提高归因于压电效应诱导的极化电场促进了光生电荷的分离,从而提高了表面反应的速率[56]。
居里温度和压电系数是反应压电材料性能的重要参数,使用温度一旦超过居里温度压电材料就会失去压电性能;压电系数反应压电材料把机械能转变成电能或把电能转变成机械能的能力,压电系数越高,能量转换效率越高。钙钛矿RbBiNb2O7的居里温度超过1000 ℃,超过了绝大多数压电材料,可在更宽的温度范围工作。有机物聚四氟乙烯(PTFE)的压电系数高达600 ×10-12 C\N,且通过超声辐照易获永久极化。Li课题组[57]将RbBiNb2O7负载在PTFE上,获得了协同效应。首先,PTFE的疏水性有利于吸附氧,压电极化分解水产生H2O2和氧,由于氧被束缚在材料表面,可通过氧还原产生H2O2。其次,PTFE与RbBiNb2O7形成异质结内建电场促进极化电荷的分离并加速迁移,提升产H2O2的能量效率。10 mg RbBiNb2O7\PTFE加入到 1 mL乙醇-水溶液中产生H2O2的速率为219.23 μmol/(g催化剂·h),换算成重量百分比为0.0074%。这个值分别是单独RbBiNb2O7和单独PTFE的12和3倍[57]。除了钙钛矿,一些金属、碳材料、有机物等也可以作为压电材料产生H2O2。
金属Pt也具有压电极化产H2O2的能力。在离子液体的帮助下,将单原子Pt锚定在二氧化硅的表面(Pt1/SiO2),组成的催化剂在超声催化下可以生成3027.1 µmol/(L·h)H2O2(重量百分比0.01%)。这是当时文献报道的超声催化产H2O2的最高浓度。H2O2的高产率归功于Pt1的高效双向催化作用:Pt1不仅具有较高的水吸附能和较低的水活化能,还可对O2选择性活化,因此可通过氧还原和水氧化两个途径同时生成H2O2[58]。
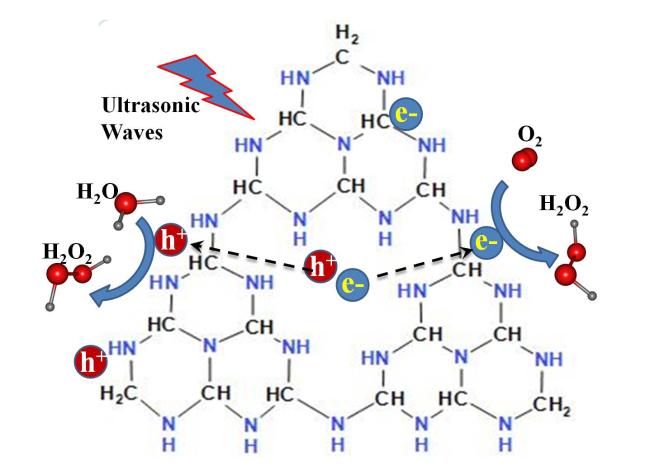
不含金属的石墨相氮化碳(g-C3N4)是一种层状二维结构,对层状材料施加外力时,层与层之间产生的应变梯度导致明显的压电响应[50]。二维g-C3N4的压电特性是由其纳米铁电性质和其晶格结构中的纳米尺度三角形非中心对称孔引起的。此外,g-C3N4具有丰富的吡啶氮是吸附和活化氧的活性位点[50]。因此g-C3N4是通过压电催化氧还原产H2O2的理想材料。上述分析得到了实验支持,50 mg催化剂放入100 mL纯水中,超声辐照下产H2O2的速率达到34 μmol/h[50],换算成重量百分比为0.0011%。尽管这个浓度距离消毒要求还很远,但这个工作为不使用金属催化剂仅以纯水和氧气为原料产生H2O2开启了新方向。超声作用与g-C3N4,通过压电效应分解水产H2O2的电荷迁移和反应路径示意图见图3 。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
有机聚合物也可作为不含金属的压电材料产生H2O2。Zhang课题组[59]研制出聚偏氟乙烯-六氟丙烯(PVDF-HFP)基体和二氧化硅纳米颗粒填料组成的新型柔性聚合物薄膜。PVDF-HFP具有弱压电效应,SiO2没有压电效应,超声辐照下单独的PVDF-HFP或二氧化硅产生的H2O2均低于10 μmol/ (L·h)。构成复合薄膜后,二氧化硅不但能强化PVDF-HFP的极化电场而且可以吸附反应中间体,在超声辐照下可产生246 μmol/(L·h)的H2O2(重量百分比0.00083%)。计算机模拟和实验结果表明,H2O2产生能力源于压电极化电场对气液固界面反应的强化作用,二氧化硅的氧原子与PVDF的氢原子构成的氢键提升了压电响应,而且二氧化硅的表面羟基对氧还原中间体-OOH的吸附能适中,有利于选择性产H2O2。这种柔性薄膜制备方法简单、环境友好、长期储存稳定、使用后易于回收,具有实际应用潜力[59]。
3.4 等离子体法产H2O2
等离子体是由电子、离子、激发态粒子和中性粒子组成的呈整体电中性的集合体。等离子体包含大量高活性粒子,能引发复杂化学反应,因此在环境、能源、材料领域应用广泛。等离子体产生H2O2有三种可能机制:等离子体中的正负电荷传输到水中分别通过氧化水或还原氧产生H2O2;气相羟基自由基合并产生H2O2随后溶解到液相;水中H2O+离子被脱氢氧化成羟基自由基后合并产生H2O2[60]。
产生等离子体可通过辉光、弧光、电晕和介质阻挡等多种放电方式。Locke课题组[61]基于弧光放电原理研制出了滑动电弧等离子体反应器,该反应器的两个曲面电极形成了渐变间隙,从2 mm逐渐扩大到20 mm。他们在两级间施加高电压击穿空气形成等离子体,然后通过喷嘴将小水滴喷射到电极之间的等离子体中,毫米级的小间隙确保水雾与等离子体充分接触,基于上一段提到的原理产生H2O2。他们使用250 Hz的脉冲电源,避免连续供电导致温度上升,防止水滴蒸发气化并抑制产生的H2O2热分解。最终实现1 mL/min进水流速产生10 mM的H2O2(重量百分比0.034%)。液相产物中H2O2浓度随进水流速增加而下降,进水流速20 mL/min 时,产生H2O2浓度为0.9 mM,此时,每kWh能量约产生80 gH2O2[61]。Ranieri等[62]的研究表明,用压缩空气携带直径0.3 μm左右的小液滴进入微秒脉冲介质阻挡放电等离子体装置,可在小液滴中产生H2O2,并均匀地覆盖在农产品的表面,完成灭菌保鲜。使用80 cm2的等离子放电截面,在1 W/cm2功率密度和50 L/min压缩空气流速条件下,完成100 L空间的消毒仅需2 min[62]。
放电时间、放电功率、液相电导等因素均可影响H2O2产率。Takeuchi等[63]系统地研究了等离子体放电产生H2O2的过程机制,发现液相中H2O2浓度随等离子体处理时间及放电电流线性增加,与放电间隙距离无关。H2O2产生位置主要在气液界面,气相中产生的H2O2和界面附近液膜中产生的H2O2通过扩散迁移到溶液中。最优条件下,在3 min内于25 mL纯水中产生约22 mg/L的H2O2,能量效率为3.9 g H2O2 /kWh[63]。H2O2的产生取决于等离子体体积和等离子体与液相的界面面积。一般来说,随着功率增加,等离子体体积膨胀,与液相的界面增加。这为水分子与电子的碰撞提供更多机会,生成更多的•OH及H2O2。液体电导率也是影响等离子体产生H2O2的关键因素。Voráč课题组[64]通过向去离子水中添加KCl获得电导率为0.01~36 mS/cm的水溶液。对于微秒脉冲等离子体,电导率由0.01 mS/cm提高到0.3 mS/cm,导致H2O2产生速率从0.12 μmol/s下降到0.045 μmol/s,降幅达62%。对于纳秒脉冲等离子,在氩等离子体中,电导率0.01 mS/cm和36 mS/cm的液膜对应的 H2O2的产生速率分别为0.16 μmol/s和0.14 μmol/s,下降约13%。说明液膜电导率提升不利于产生H2O2,且脉冲频率越低这种不利影响越强[64]。这些工作确认了在导电溶液中等离子体放电也能产生H2O2,因此表明这种方法可用于海水或其他电性水灭菌。
等离子体背景下,产生H2O2的位置分为气相和液相。为研究不同位置对产生H2O2的贡献,Li课题组[65]对比了水面和水下鼓泡脉冲放电产生H2O2的浓度,无论放电模式如何,H2O2的产率和生产速率随电压的增加而增加,且在200 mL纯水中鼓泡脉冲放电产生的H2O2速率为0.0196 mg/min,明显高于水面脉冲放电的0.012 mg/min。考虑到鼓泡过程在液膜内形成了更多的气液界面,这一结果表明气相中更容易形成H2O2[65]。Tachibana等[66]考察了类似的鼓泡放电体系和水面放电体系的单位能耗H2O2产量,放电10 min,鼓泡体系在12 mL水中产生270 mg/L的H2O2,每kWh电能产生1 gH2O2,水面放电体系则在15 mL水中产生约93 mg/L的H2O2,每kWh电能产生0.1 gH2O2。这个结果进一步说明等离子体方法在气相中产生H2O2更经济[66]。等离子体要求毫米尺度的放电间隙,如何大规模产生一直是该技术的研究热点。Vlachos课题组[67]研制了一种可用于分布式生产H2O2的新型等离子体反应器,该反应器基于介质阻挡放电原理,特点是填充了高密度的气液界面。可在4 kV电压和0.7 W的功率下运行,产生2.2 mM的H2O2,提高功率可进一步获得浓度为33 mM的H2O2,使该技术易于通过单元反应器串并联的方式扩增,有希望实现H2O2的规模化生产[67]。
3.5 微液滴法产H2O2
为了制造能够模拟微液滴过程的固液界面,Chen等[68]使用聚二甲基硅氧烷在玻璃上构建出密闭的直通道(直径20 μm,长度100 μm),通入去离子水,即可检测到H2O2。在这一过程中,生成H2O2所需的氧原子可能来自细颗粒表面的含氧基团。为了验证这个想法,以二氧化硅为衬底,用O2等离子体活化衬底增加羟基密度,结果表明羟基密度与固液界面产生H2O2的量正相关。同位素实验证实H2O2中的氧原子来源于衬底的羟基,使用含有羟基自由基清除剂的水制作的微液滴就很难产出H2O2。分析表明,细颗粒接触水时,离子转移、电子云的重叠和表面羟基变化等都可引起表面电荷转移,引发固体表面羟基重组产生H2O2[69]。这种微液滴产生的H2O2的方法仅以水为原料,且无需能量输入,是值得研究的新方法。
为了强化空化现象,增加H2O2产量,在超声雾化器上引入锌片[74]。与均匀液相中的空化相比,锌片上存在的表面缺陷,如裂纹,增加了空化气泡的成核位点。反过来,空化气泡崩溃形成的微射流不断冲击和研磨锌片,进一步增加了表面缺陷(裂纹、孔洞、表面粗糙度等)的数量,从而暴露出更多的活跃的位点。成核位点的增多可以提高空化效果。因此,锌片的腐蚀和H2O2的生成可以互相促进。该方法产生H2O2的反应式如下。
Zn−2e−→Zn2+
e−+O2→∙O2−
e−+∙O2−+2H+→H2O2
Zn2++2OH−→Zn(OH)2
4 结论与展望
本文介绍了通过光、电、热、声、等离子、微生物代谢等方式提供能量实现主要以水为原料原位产生H2O2的新方法。表1 对比了各种技术产生H2O2的典型浓度和一般产量,由于这些方法使用了不同的关键材料和设备,在H2O2浓度、产量方面存在差异。可以看出,浓度和产量两个角度都距离实际消毒要求有一定差异。
表1 以水为原料现场产H2O2方法的对比Table 1 Performance comparison of in-situ H2O2 preparation via water oxdation |
此外,现有报道中对于各种方法产单位重量H2O2的设备成本、运行能耗、关键材料寿命的数据较少。因此尽管这些方法有很多思路和方法值得我们借鉴,但还难以满足实际消毒的要求,存在的主要问题及对策如下:
第一,绝大部分报道的H2O2积累浓度没有达到消毒要求的3%。解决这个问题关键是研制高效催化材料,新材料应具备H2O2选择性,能抑制H2O2生成过程的竞争反应。此外,需要结合理论计算和实验研究明确H2O2的自分解条件和自分解浓度下限,在制备H2O2过程中回避这些条件。如果在制备H2O2条件下H2O2的理论积累浓度存在上限且不满足消毒要求,应开展原位浓缩方法研究,建立后处理工序提升浓度而不是一定在制备条件下追求高浓度。
第二,报道的工作产生高浓度H2O2的速度慢能耗高,单位体积反应器每小时产生的H2O2溶液体积通常只有几十毫升,单位电能输入产生的H2O2不超过1 g,成本远高于工业集中制备的市售产品,这些均难以应对实际消毒应用。新材料、反应器 效率、反应机制等均对解决这个问题具有重要 意义。建立关键材料对OOH基团的吸附能调控方法,设计关键材料高密度填充的单体反应器,模仿自然界光反应、生物过程探索产H2O2高效机制。
第三,报道的新材料和实验体系体积小、寿命短,面向应用的技术需要长寿命材料和可规模化的材料制备方法。建议从原料来源和成本、制备工艺、生命周期等方面提前筛选新材料,建立可批量制备的技术路线;不能忽视具有氧化性的H2O2对关键材料的腐蚀作用,应采取措施加速产生的H2O2脱离反应位,提高材料寿命;研制单元反应器的串联或并联方法、材料面积或体积的扩大方法,设计总体积与实际应用相当的反应装置,考察实用性,发现应用过程存在的问题并探索解决办法。