





1 引言

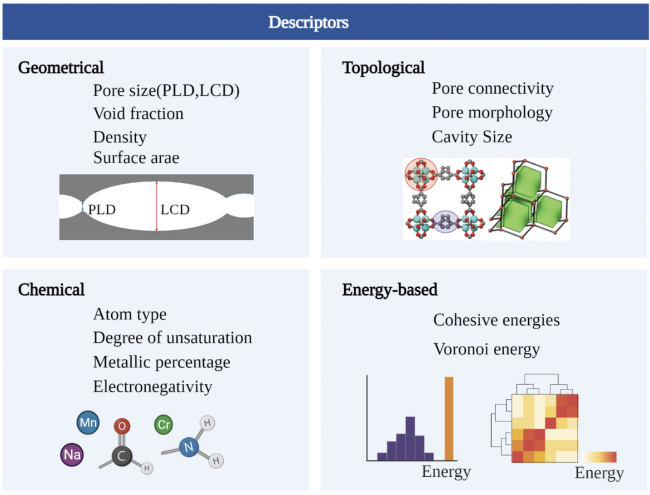
2 方法

2.1 高通量筛选技术
2.1.1 MOFs的蒙特卡洛模拟
2.1.2 分子动力学模拟
2.2 机器学习方法
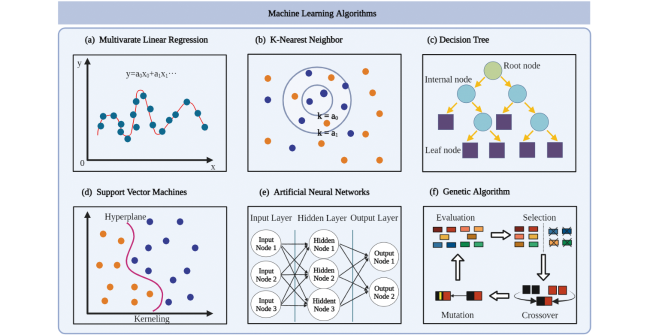
2.2.1 多元线性回归
2.2.2 K近邻
2.2.3 决策树
2.2.4 随机森林
2.2.5 支持向量机
2.2.6 神经网络
2.2.7 进化算法
2.2.8 模型评估参数
3 研究
3.1 气体储存
3.1.1 H2储存
表1 机器学习预测MOFs储氢性能Table 1 The H2 storage performance of MOFs predicted by ML |
| Researcher | Descriptors | Algorithms | Best Results |
|---|---|---|---|
| Cao[71] | Geometrical + Chemical | LSSVM, ANFIS, ANN | R2 = 0.990, MAE = 0.050 wt%, MSE = 0.059 wt% |
| Bucior[73] | Energy-based | LASSO | R2 = 0.960, MAE = 2.40 g/L, RMSE = 3.10 g/L |
| Giappa[74] | Energy-based | KRR, RFR, SVR | MAE = 1.160×10-4 Ha |
| Ahmed[75,76] | Geometrical | DT, RF, LR, SVM, ERT, etc. | R2 = 0.997, MAE = 0.10 wt%, RMSE = 0.180 wt% |
3.1.2 CH4储存
表2 机器学习预测MOFs储甲烷性能Table 2 The CH4 storage performance of MOFs predicted by ML |
| Researcher | Descriptors | Algorithms | Best Results |
|---|---|---|---|
| Pardakhti[79] | Geometrical + Chemical | RF | R2 = 0.980, MAPE = 7.180 cm3/g |
| Wang[80] | Geometrical | Graph Convolution Neural Networks (GCNN) | AUC = 0.978 cm3/ cm3 |
| Beauregard[78] | Geometrical + Chemical | RF, GA | R2 = 0.950, MAPE = 1.370 cm3/g |
| Fanourgakis[81] | Geometrical + Topological | RF | R2 = 0.990, RMSE = 9.050 cm3/g |
| Kim[82] | Geometrical + Chemical | SVM, DT, RF | R2 = 0.947, MSE = 0.918 kJ/mol, MAPE = 3.903 kJ/mol |
| Gurnani[19] | Geometrical + Chemical | Feed-forward Neural Networks | R2 = 0.990, RMSE = 7.830 cm3/g |
| Suyetin[20] | Geometrical | MLR | R2 = 0.990 |
| Lee[83] | Topological | MSGA-FA | * |
| Taw[84] | Geometrical + Chemical | BO | R2 = 0.960 |
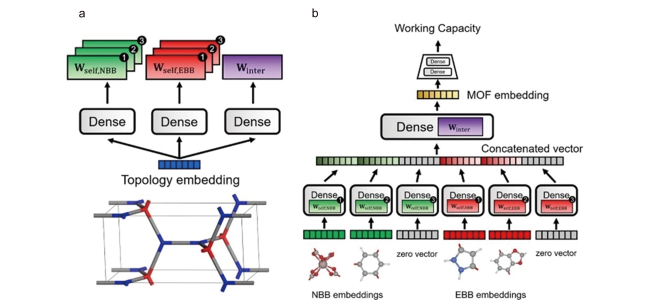
图5 MOF-NET模型图。“Dense”代表全连接层。(a)拓扑嵌入与拓扑嵌入权重的产生。Wself,NBB代表NBBs自身相互作用产生的权重,Wself,EBB代表EBBs自身相互作用产生的权重,Winter 代表相互作用产生的权重。(b)MOF-NET模型主要组成部分。模型由简单的全连接层组成,但某些层的权重由拓扑嵌入[83]Fig. 5 Schematics of the MOF-NET architecture. The word “Dense” refers to fully connected layers. (a) Topology embedding and the generated weights from topology embedding. Wself,NBB is the self-interaction weight for NBBs, Wself,EBB is the self-interaction weight for EBBs and Winter is the interaction weight. (b) Main architecture of MOF-NET. The MOF-NET consists of simple fully connected layers, but the weights of some layers are generated from the topology embedding[83] |
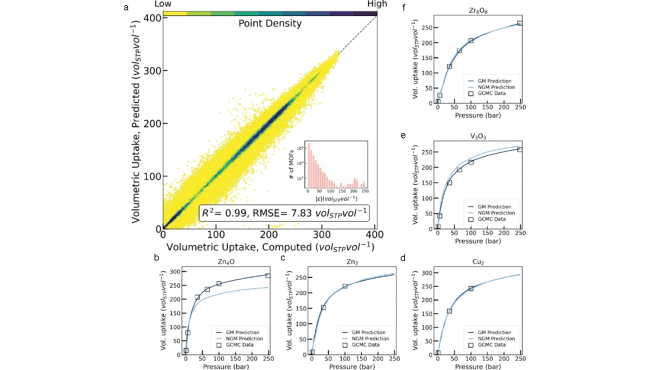
图6 (a) GM模型预测的533 430种CH4体积吸附量结果;(b~f) 示例结构中GM、NGM与GCMC预测吸附曲线结果对比[19]Fig. 6 (a) Computed volumetric methane uptake vs predicted volumetric methane uptake for each of 533 430 measurements in the curated hMOFs data set;(b~f) NGM and GM predicted isotherms (light-blue and dark-blue curves, respectively) vs GCMC data (white squares) for five randomly selected MOFs[19] |
3.1.3 碳捕捉
表3 机器学习预测MOFs的分离性能Table 3 The separation performance predicted by ML algorithms |
| Separation system | Researcher | Algorithms | Descriptors | |
|---|---|---|---|---|
| Noble gas | Xe/Kr | Liang[92] | Ridge, LASSO, Elastic NET, SVM, Bayesian, ANN, RF, XGB | Geometrical |
| Xe/Kr | Ma[93] | DNN | Geometrical | |
| Ar/Xe/Kr | Anderson[94] | DNN | Geometrical + Topological | |
| Xe/Kr | Liu[95] | BPNN | Chemical | |
| Cx/Cy | ethane/ethylene | Halder[96] | RF | Geometrical + Chemical |
| ethane/ethylene | Wu[97] | LR, DT, RF, SVM, KNN, GBT | Geometrical + Chemical | |
| C4/C7 | Tang[98] | MRGP | Geometrical + Chemical | |
| p-xylene/m-xylene/o-xylene | Qiao[99] | BPNN, DT | Geometrical | |
| Other | CH4/H2S | Cho[100] | RF | Geometrical + Chemical |
| N2/O2 | Yan[101] | RF, GBT, XGB | Geometrical + Chemical | |
| D2/H2 | Zhou[102] | SVM, RF, GBT, DNN | Geometrical + Chemical | |
3.2 气体分离
3.2.1 稀有气体分离
表4 机器学习预测MOFs储二氧化碳性能Table 4 The CO2 capture performance predicted by ML |
| Researcher | Descriptors | Algorithms | Results |
|---|---|---|---|
| Burner[21] | Geometrical + Topological | DNN | R2 = 0.975, RMSE = 10.0 mmol/g |
| Dashti[86] | Geometrical + Chemical | PSO-ANFIS, RBF-ANN, DE-ANFIS, LSSVM | R2 = 0.997, MSE = 0.167 mmol/g |
| Deng[22] | Geometrical + Chemical | BPNN, RF, DT, SVM | R2 = 0.994 |
| Zhang[87] | Chemical + Topological | RNN, MCTS | * |
| Li[88] | Chemical | SVM, KNN, DT, SGD, NN | R2 = 0.940 |
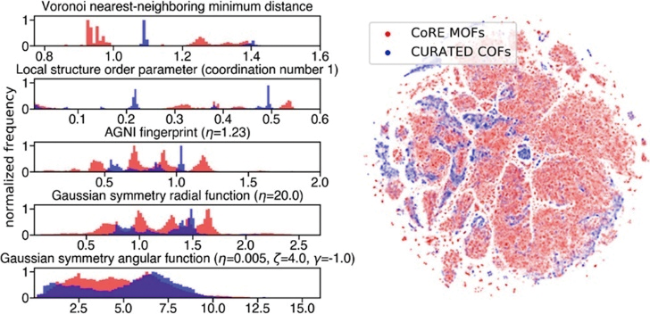
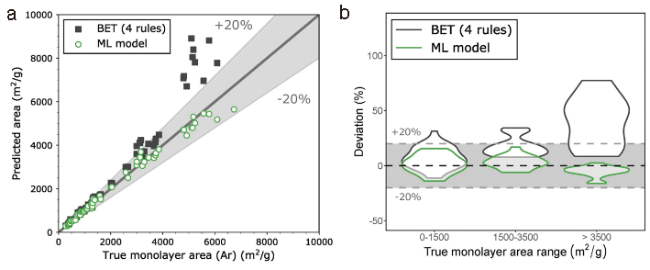
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}