



1 引言
在常规的高分子合成过程中,液态溶剂作为反应介质往往是不可或缺的重要组成部分。除本体聚合反应(不需要溶剂)发生在本体内,其他聚合反应的发生场所一般都在溶液内(溶液聚合)、液滴内(悬浮聚合)、胶束和乳胶粒内(乳液聚合)。相比于无溶剂的聚合体系,溶剂是一种非常好的热传导介质,可调控、降低聚合反应温度,避免单体和产物的分解;另外,聚合物反应过程中产生的小分子可以通过与溶剂的共沸或反应等方式被脱除,使反应变得平稳易控。因此,液态溶剂是一直伴随高分子材料科学发展的重要研究对象,在液态溶剂下发生的与高分子凝聚态相关的化学问题已经得到深入和广泛研究,如反应类型、反应机理、反应动力学、聚合产物形态结构的生成与演变等。
熔融缩聚(1930s)[6⇓~8]、反应挤出(1960s)[9⇓⇓~12]、固相缩聚(1960s)[13⇓~15]就是这些无溶剂树脂合成技术中的典型代表。相比于在溶液中发生聚合反应的常规树脂生产技术,在反应过程中不使用溶剂是这三种技术最大的共同特点和优势,而且在聚合反应完成后的粗产物中副产物和杂质的含量也较少,能够省去繁琐的纯化分离步骤,使生产成本大幅降低,同时还能够达到节能、减排的目的。例如,采用传统生产工艺制备15万吨SBS(苯乙烯-丁二烯-苯乙烯嵌段共聚物)需要使用60万吨有机溶剂。如果打算回收用于生产的全部溶剂, 则需要消耗~240亿千卡能量,这相当于燃烧~280万立方米的天然气或~280万kW·h 电所产生的能量。但如采用反应挤出加工技术生产SBS, 不但可以避免因使用溶剂而带来的高能耗和环境污染等系列问题,还符合“碳中和”的全球可持续性发展战略要求[5]。
在无溶剂下发生的聚合反应大多属于放热反应,由于没有溶剂作为传热介质,当聚合反应达到一定程度后,物料的温度会急剧上升,在数分钟内会达到300 ℃以上,生成的相应聚合物易发生降解和碳化,因此必须将反应过程中产生的聚合热及时移除。为解决这个关键问题,获得更高品质的高分子树脂,除需要开展化学工程和工程热物理学科的基础问题和新技术研究工作外,更应该关注并深入研究无溶剂下的制备聚合物的化学反应类型、反应机理及相关反应动力学等问题,这些问题又都是凝聚态变化下的化学问题。例如,熔融缩聚、反应挤出、固相缩聚这三种聚合技术在生产高分子树脂过程中物料的凝聚态变化一直与聚合期间所发生的化学反应密切相关,针对凝聚态变化与化学反应的相互作用和影响关系进行研究,就能更好地对相应生产技术的发展提供理论指导,促进无溶剂生产技术的突破创新。本文将通过总结前人的工作和相关经验,对这三种聚合方法和机理进行简要的分析介绍,并通过一些典型的高分子树脂生产实例进一步理顺三者之间的相互联系,揭示这三种无溶剂型高分子制备技术过程中出现的高分子凝聚态变化与化学反应之间的基本问题,为广大科研工作和提供一些有价值的参考。
2 无溶剂参与的三种典型高分子树脂生产技术简述
本文将按照熔融缩聚(熔融态)、反应挤出(熔融态)、固态缩聚(固态)的顺序对三种聚合方法进行逐一的简要介绍,然后再一并综合比较和分析彼此间的差异和联系,展示这三种聚合方法发展的相关性。
2.1 熔融缩聚技术
1908年,聚酰亚胺(PI)首先成为能够通过熔融自缩聚反应制备的合成树脂材料,但此时熔融缩聚法还没有得到人们的广泛关注[6]。直到第二次世界大战爆发,为满足聚酰胺类材料在军事方面的巨大应用需求,熔融缩聚技术才正式登上合成树脂的历史舞台,向人们展示了其简单高效的生产能力。随后不久这项技术就开始被广泛应用于聚酯、不饱和聚酯、聚胺酯等更多高分子材料的实际工业化生产中,并成为树脂生产和技术研究领域的热点,例如,对苯二甲酸乙二醇酯的缩聚(PET聚酯,涤纶)[16]、对苯二甲酸丁二醇酯的缩聚(PBT聚酯)[17,18]、己内酰胺的缩聚(尼龙6,锦纶)[19]等高分子树脂都可以通过熔融缩聚技术实现工业化生产,目前单套熔融缩聚装置的生产能力已经达到数万吨/年以上。
熔融缩聚的优点是:(1)产品纯度高:由于熔融缩聚不用溶剂,因此反应物浓度高、反应速率快、引入杂质机会少,所以产品质量较均一,得到的产物纯净、分子量分布窄,不需要额外的产品提纯工艺;(2)生产流程简单:由于熔融聚合不用溶剂,免除了操作复杂且耗能高的溶剂分离工艺,生产流程大大简化;(3)易于大型化生产:由于没有溶剂,反应设备容积率高,所以生产能力大;(4)便于连续化生产:缩聚反应完成后的产品仍处于熔融状态因此聚合物熔体可以直接纺丝、成膜或铸带切粒,减少了能耗,缩短了加工工艺过程,提高了生产效率。
但缺点也很明显:主要是分子量不够高。导致这一结果的原因是:(1)在熔融缩聚过程中,随着聚合度的增加,釜中反应体系(熔融态)的黏度急剧增加,导致同时期反应生成的低分子副产物难于从高黏度的反应体系中排除,随着缩聚反应时间的延长,这些无法排除的副产物又导致聚合物的降解反应加剧;(2)反应后期,聚合程度增大导致反应体系黏度随之增大,反应体系内的传热变得更加困难,容易出现局部过热的问题,导致降解反应增加,也限制了分子量的进一步提高;(3)在聚合反应釜中,高黏度流体易处于“死区”,带来严重的黏釜现象,高温下的较长时间停留会引起降解等各类副反应发生,使产品的色泽加深,质量变差,出料时也更加困难且容易碳化变黑。一般认为解决上述问题的关键在于对反应器设计的优化和改良,只要保证缩聚反应器内熔融体系的流动和混合状态好、传热传质效率高、过程满足平推流,就有望获得分子量高且品质均一的树脂产品。
2.2 固相缩聚技术
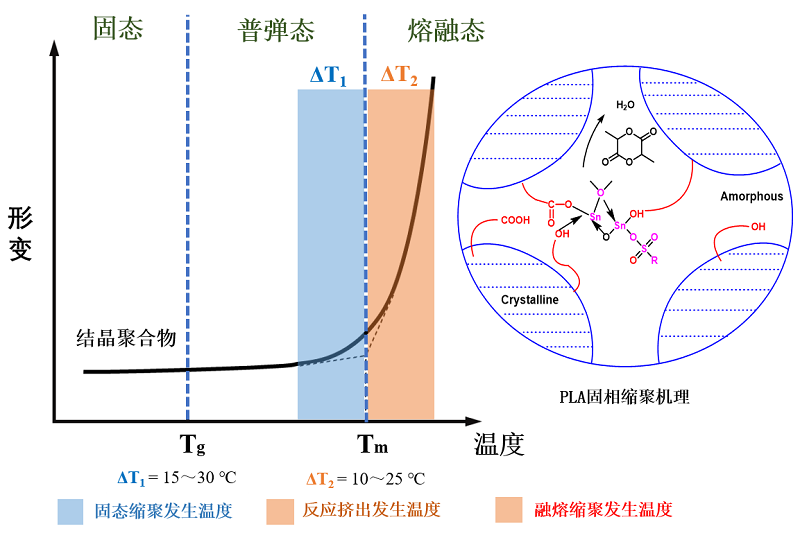
20世纪30年代末,美国科学家Paul[15]在研究尼龙的熔融缩聚法时发现:通过熔融缩聚得到的尼龙在冷却到固态后,如果将其维持在一定温度,则这些尼龙分子之间仍然可以继续发生缩聚反应,成长为具有更高分子量的长链尼龙聚合物。由于这一反应发生时所处的温度在尼龙的熔点之下、玻璃化温度之上,所以在缩聚反应进行时反应物宏观上仍呈现固体状态(处于结晶聚合物的普弹态),所以这个反应被形象地命名为固相缩聚(SSP)。但在那个时代,基于对熔融缩聚的认识和理解,人们往往认为固态下的单体分子都是相对冻结的,彼此间缺乏碰撞机会,都难以进行聚合反应,更何况是具有一定分子质量的聚合物,所以这一工作并未引发太大的重视。1945年一则关于采用辐射引发固态丙烯酰胺聚合,成功得到高分子量聚丙烯酰胺的工作被报道后,关于“固态聚合过程”的研究才开始在高分子科学界得到一定程度的重视[23]。从到20世纪50年代后期到60年代,工业上开始采用这种方法制备高分子量的尼龙、聚酯等缩聚型高分子材料[20,24⇓~26],推动了涵盖固相缩聚反应过程的控制方法、反应机理、物理化学等内容的系统研究工作的开展,并大量报道。20世纪70年代,美国杜邦公司成功地采用固相缩聚(也可被称作固态缩聚或固相增黏)技术研制出瓶级聚酯切片,并投入工业化生产,以此技术生产出的聚酯重量轻、强度高、透气性和阻气性好,使得固相缩聚技术在产业界的应用得到了迅速的发展[8]。同一时期,Wegner[27]率先采用固态聚合法(Solid-phase polymerization)成功制备出聚双炔类的宏观单晶体, 为研究聚合物结构与性能之间的关系提供了理想的一维晶体模型,并让人们了解到固态聚合法具有反应活化能低、纯度高、成本低、无诱导期,且有明显的聚合反应后效应和聚合前后分子结构受局部规整性控制等特点,自此针对固态聚合的研究成为了科学界和产业界的热点,涵盖固相缩聚法的“固态聚合”也成为了固态化学的重要分支。经过不断发展,固态聚合已经可以采用更多的引发方式引发,如高能辐射或紫外线、热和电极等,而聚合反应机理也不再局限于最初的逐步聚合(缩聚),进而拓展到具有自由基和离子链式反应特点的加成聚合,可制备的聚合物种类不断扩展。
目前公认的固相缩聚原理是:当温度在聚合物玻璃化温度以上、熔点以下时,虽然聚合物大分子链被固定,但分子链末端基具有一定的活性,能够通过扩散互相靠近发生反应,生成的小分子被真空或气流带出,使得反应能够继续进行;所以,固相缩聚反应一般会经历以下4个阶段:(1)活性基团的互相靠近与碰撞;(2)官能团之间发生可逆化学反应;(3)生成的小分子副产物从内部向粒子表面扩散;(4)小分子从表面向惰性气体或真空中扩散脱离体系。相关机理、聚合反应动力学及其影响因素方面研究的报道也越来越多。
由于缩聚反应是在固态下进行的(属于均相缩聚),因此这是一种处于固态下实现分子量提升的高分子树脂制备技术。因该技术能够使聚合物切片(如PET切片)在固相状态下进行缩聚反应,从而达到聚合物黏度上升(宏观上)、分子量提高的目的,所以也被形象地称为“固相增黏”技术。
相比于熔融缩聚技术,固相缩聚技术具有以下优点:(1)反应温度较低,所以能够有效减少聚合过程中发生的降解反应和副反应,避免出现产物变色或分子量降低的产品质量问题;(2)能够显著提升聚合物的相对分子质量,使其物理机械性能与成型加工性能都得到改善,进而拓宽产品的应用领域;(3)由于固相缩聚反应过程无需搅拌装置,因此反应釜结构简单,避免了熔融缩聚后期出现的高黏度聚合物难以搅拌和出料困难的现象,且由于所需反应温度低,所以能够有效降低生产所需的能耗;(4)固相缩聚法所需反应条件温和,并且全程不使用液态原料。这样既减少了回收溶剂的工艺,节约了生产成本,而且避免了液态原料泄露对环境产生污染;(5)固相缩聚反应工艺相对简单、灵活,既可以选择连续化操作,也可以采用间歇式操作。
但是固相缩聚技术自身也同样存在一些固有的缺点:(1)反应速率较慢,消耗的反应时间很长;固相缩聚的反应活化能远远高于熔融缩聚,但反应温度却比聚合物的熔点还低,因此反应物处于固体状态,活动空间有限,并且分子链端基获得的能量较少,反应活性降低,导致扩散速度和反应速率均较慢。(2)固相缩聚增效效果好,但重现性差,产品质量不稳定。(3)敞开体系的固相缩聚,利用源源不断的惰性气流将小分子副产物带离体系,虽能有效促进反应向正反应方向进行,但会增加生产成本且消耗大量的资源;而密闭体系的固相缩聚虽节约能耗,操作简单,但会限制缩聚反应平衡,无法有效提升聚合物的相对分子质量。
2.3 反应挤出技术
这一技术诞生于20世纪50年代,最初被视为一种新型的混合技术[10,12,28];直到20世纪60年代后期才首先在欧、美、日等发达国家和地区被应用于聚合物的合成和聚合物改性方面的研究[31⇓~33],尤其是在制备开发新型高分子材料方面更是效果显著,因此各大研究机构和挤出设备的生产厂家都高度关注该技术,使其迅速发展起来,目前反应挤出技术已经成为国际上关注度较高的研究方向。德国Achen大学的Menges、美国Akron大学的White及英国Brumel大学的Homsky等分别在1986年和1994年发表的文章中较全面地剖析了尼龙-6双螺杆反应挤出合成中各种加工参数及条件因素之间的影响情况,将反应挤出尼龙-6的研究开发工作推向新的高度。美国科学家Waymouth通过控制聚烯烃的微观结构,利用连续反应成型技术,直接由丙烯单体制备了热塑性弹性体纤维。美国Bodolus报道生产丁腈橡胶时,丙烯酸酯与丙烯睛在挤出机中的共聚反应,制得的共聚物的冲击强度比一般丙烯酸酯——丙烯睛共聚物高10倍。直到20世纪80年代初,这一技术才真正走进了我国的学术界和工业界工作者的视线中,人们开始逐渐着手从事该领域的研究与探讨。经过科研工作者的坚持和不懈的努力,反应性挤出加工也广泛地应用在聚合物合金的制备、接枝、偶联/交联、降解等方面[34],并且在反应挤出技术的某些方面我国也已达到世界先进水平。
与传统的聚合釜式生产需要经过聚合、分离、纯化以及再挤出造粒成型等工艺技术相比,反应挤出本体聚合技术综合了原料的连续本体聚合以及聚合产物的熔融成型为一体,充分利用螺杆挤出机中有限的停留时间使聚合反应“一步到位”得到所需要的产物,因此具有明显的技术优势[2,5,29,35,36]:(1)反应过程可连续进行,通过调整温度设置、螺杆转速和加料位置,实现对反应过程的精确控制,调控聚合反应开始与停止的最佳时间;(2)可以不用或少量使用溶剂,聚合后无需增加分离操作,不但节约能源,而且可以减少环境污染;(3)通过调节挤出机筒体和螺纹块结构与阻隔圆环等组合,可以控制停留时间、反应温度及物料分布等,有利于获得分子量分布窄、性能均一的产品;(4)可灵活调整螺杆结构及挤出工艺,以适应不同的物料体系,具有生产多种产品的灵活性;(5)既能控制物理结构,又能通过聚合控制化学结构,得到特异性能的新型聚合物;(6)可以实现常规反应器中难以操作或不能实现的反应过程,尤其适用于反应速率快、黏度高、热效应较大的体系,以及反应过程中涉及到固液相变的反应过程;(7)在同一条生产线上同时实现聚合反应(或改性)和成型(造粒或得到最终产品),既简化工艺流程,又降低生产成本。此外通过改变螺纹块结构,变更物料的流变行为可以按预定的要求使产物的物理织态发生演变,从而取得传统方法不能得到的高性能聚合物以及特殊分子链结构的聚合物。因为反应挤出技术具有较高的性价比及比较显著的社会经济效益,必然成为最为经济和最有前途的聚合方法之一。
当然,反应挤出技术也存在一些问题尚待解决。(1)技术难度大:针对不同的材料不但要进行配方和工艺条件的研究,还需要针对不同的反应设计相应的新型反应挤出机,所以对于新的体系常常存在研发资金投入大、研发时间过长的问题。(2)反应过程中难以观察检测:参与反应的物料在挤出机进行挤出反应时,始终处于动态、封闭的高温、高压环境内,操作者无法通过观察检测物料的状态变化判断具体的反应程度; 挤出过程中物料在料筒中的停留时间比较短(一般只有几分钟), 但却要求料筒内的化学反应必须快速完成,如果反应超过20 min,就会引发更多副反应,丧失了反应挤出技术的应用意义。(3)技术含量高:反应挤出技术涉及到高分子材料、高分子物理、高分子化学、化学工程、聚合反应工程、橡塑机械、聚合物成型加工、机械加工、电子、材料等诸多学科, 要取得一定的研究成果必然需要经历较长的研究,并得到多领域的支持合作。综上所述, 反应挤出技术具有研发投入高、技术含量高、产品利润高的特点,虽然在研发阶段遇到的困难比较多,但在工业应用上优势明显。
3 无溶剂树脂生产技术的典型应用
我们选取具有代表性的聚对苯二甲酸乙二醇酯(Polythylene terephthalate,PET)和聚乳酸(PLA)树脂为例,将三种技术在实际生产中的应用情况和现存的问题进行介绍和分析。
3.1 PET树脂的无溶剂生产
3.1.1 PET简介
PET俗称涤纶,是一种高度结晶性聚合物,多数呈乳白色或浅黄色,表面平滑而有光泽,其结构式如图1 所示。其玻璃化转变温度(Tg)为 83 ℃,熔融温度(Tm)约为255~265 ℃,可在120 ℃温度范围内长时间使用。由于对气、水、油及异味具有较强的阻隔性,透明度高、光泽性好、安全性好,且价格相对便宜,被广泛应用在纤维制造、薄膜生产、瓶用包装以及工程塑料领域,全球市场的需求量巨大。尽管PET的世界年产量逐年高速增长,却一直无法满足日益增长的应用需求。提高PET生产能力、提升PET树脂黏度、开发PET回收加工再利用技术,成为当下PET树脂生产开发领域的研究重点。

3.1.2 PET树脂的工业化合成路线
目前,世界各国生产PET树脂所采用的技术路线主要是酯交换法(也称DMT法)和直接酯化法(PTA法)两种。DMT法是通过对苯二甲酸二甲酯(DMT)与乙二醇(EG)进行酯交换反应,然后缩聚成为PET;而PTA法则是采用高纯度的对苯二甲酸或中纯度对苯二甲酸(MTA)与EG直接酯化,然后缩聚而成。上述两种方法在实际生产过程中各有利弊,但自从高纯度PTA的重结晶方法实现工业化生产之后,采用PTA技术的生产成本大幅降低,其自身优势相比于酯交换法变得更加突出。目前PTA法已经凭借着原料消耗低、EG回收系统较小、无副产物甲醇、反应速度平缓、生产流程较短、生产控制比较稳定、生产安全性较高、工程投资低及生产成本更低等诸多优势,成为了全球PET生产应用最多的主流方法。
3.1.3 典型的PET熔融缩聚生产过程
以PET聚酯的熔融缩聚生产过程为例,其过程主要有以下两步反应:第一步反应是PTA与EG之间进行的酯化反应,用以生成预缩聚原料对苯二甲酸乙二酯(BHET)和少量的;第二步是BHET在催化剂的作用下发生缩聚反应生成PET树脂。具体流程为:首先是将固体TPA、聚合催化剂(锑系化合物)与液态的EG三者混合制浆,其中EG与TPA的摩尔比为2∶1。制浆后进入酯化反应阶段,为缩短反应时间,此时釜内的反应压力大于大气压力(0.05~0.2 MPa),反应温度也高于EG的沸点温度(258~263 ℃)。观察这一阶段体系内凝聚态的状态变化情况可以发现,固体TPA在该反应条件下只能部分溶解于液态的EG中,因此,在酯化反应过程的前期,釜内的反应体系为“固-液非均相”体系,此时的酯化反应首先发生在因扩散作用而溶解的TPA与EG间,进行的是均相酯化反应,并开始不断脱出水、生成BHET;随着反应的进行,BHET的含量越来越高,由于TPA在BHET中具有较好的溶解性,所以TPA开始在EG/BHET(及少量短链低聚物的预聚体)混合体系中溶解,而且溶解速度随着低聚物的增加而增大,体系逐渐由非均相转向均相,由混浊趋向透明,TPA全部溶解时釜内的混合体系出现“清晰点”,标志着固相TPA消失,开始均相反应阶段。实验结果表明“清晰点”一般出现在酯化率达到85%左右时。随后,生产过程开始由酯化向第二步缩聚反应过渡。第二步发生的是BHET的缩聚反应,属于聚酯合成中的链增长反应,两个β-羟基乙酯基之间反生缩聚,脱去一分子的EG,反应式如下。该阶段缩聚反应所处的反应温度须高于PET树脂的熔融温度(Tm = 245~260 ℃),但同时也要求温度低于300 ℃(PET树脂在此温度之上开始出现降解),因此PET的熔融缩聚反应最合适的温度范围是275~290 ℃(经验上一般选择熔点之上20~30 ℃)。缩聚过程的反应时间至少为2 h,具体视反应器不同而有所不同。这个反应的反应常数较小,因此在反应过程中还须尽快地除去反应所生成的乙二醇,打破反应平衡,促使反应继续向右进行,否则不但会影响反应速度,而且聚合度也不会提高。因此缩聚要求在真空下进行,特别是缩聚后期要求在高真空度下进行,同时应尽量增加蒸发表面。
由于熔融缩聚的反应温度在聚酯熔点以上,是没有任何介质的本体聚合,所以得到的产物纯净,不需要分离介质,生产成本较低,适合简单种类、大型持续化的生产过程。另外,聚酯熔融缩聚后出料的状态一般为聚酯熔体,因此只要聚酯熔体的性能和质量达到要求就可以直接进行加工成型,非常经济便利。但是,通过熔融缩聚技术很难得到高特性黏数的PET聚酯,因为熔融缩聚过程中,随着反应体系黏度增加,反应生成的低分子副产物的排除变得更加困难,同时容易发生反应体系的局部过热现象,就导致降解反应增加,限制分子量进一步提高,所以只能生产一些低特性黏度的PET树脂。因此,要获得更高分子量的树脂材料,需要开展进一步的聚合物增黏处理。
3.1.4 PET的增黏方法
研究表明,PET聚酯的性能受其分子量影响巨大。PET树脂的分子量增大,体现为特性黏度值增高,其力学性能也随之提高。如表1 所示,聚酯的分子量大小直接决定了其的不同应用领域。普通分子量的PET树脂(特性黏度[η]为0.55~0.64 dL/g)可用于制造常规涤纶纤维,而高分子量PET聚酯则可用于制造聚酯瓶、工程塑料,超高分子量PET聚酯可用于制造结晶聚酯、缆绳(如轮胎帘子线)、编织物(涂层织物、帆布、过滤布等)、增强材料、石棉代用品、保护用品(如防弹背心)、体育用品(网球拍、钓鱼杆、球棒、赛艇等)。
表1 不同特性黏度指数PET的作用Table 1 The applications of PETs with various intrinsic viscosity |
| [η]/dL/g | 数均分子量 | 用途 |
|---|---|---|
| 0.55~0.64 | 19000~22000 | 纺织涤纶纤维 |
| 0.64~0.68 | 22000~24000 | 薄膜 |
| 0.68~0.74 | 24000~26000 | 工业丝 |
| 0.74~0.90 | 26000~34000 | 瓶、工程塑料、易拉罐、拉链 |
| 0.84~1.10 | 31000~39000 | 超强工业丝线 |
| 1.04 | 37000 | 结晶聚酯 |
(1)PET熔融增黏技术
通过熔融缩聚增黏法是指通过延长熔融缩聚的时间提升聚合程度的方法[8,42]。其聚合生产装置与普通的熔融聚合装置相比,最大的区别是:在常规熔融缩聚的终缩聚釜之后,再增加一个缩聚釜,目的是通过延长熔融缩聚反应时间提高分子量。其中,前缩聚釜中的反应温度通常为270 ℃﹐后缩聚釜中反应温度为270~280 ℃﹐在进行终聚过程中还会加入少量稳定剂以提高溶体的热稳定性。另外,缩聚反应还必须在高真空(余压不大于266 Pa)及强烈搅拌下进行﹐才能获得较高分子量的聚酯。但是一般产品的特性黏数也不超过0.9 dL/g,且产品质量也不高。另外,由于经此长时间熔融缩聚生产的高黏度聚酯中乙醛含量偏高,所以不适于生产瓶用聚酯。
总的来说,采用熔融缩聚生产高特性黏度PET树脂的成本较低,适合单一品种、大型连续化生产过程;但产品特性黏数的提升有限,产品质量差,乙醛含量偏高,不适用于生产瓶用PET。所以自20世纪80年代以后,这种工艺已经较少采用,目前使用这种工艺的大公司只有美国DuPont和Allied公司。
(2)PET固相增黏技术

例如,将分子量较低的PET预聚物(切片)加热,待温度介于熔点和玻璃化温度之间时,通过抽真空或通入保护气来增加PET聚合度。在该方法中通常使用特性黏度为 0.70~0.81 dL/g的小瓶级 PET,并在210 ℃冷凝15~20 h,才能获得高黏度、高品质的PET树脂。由于固相缩聚反应温度低、速度慢、工艺流程长,所以投入大规模生产将会有较高的成本。
在目前国内外的工业生产中,固相聚合分为间歇式和连续式(表2 ),两种工艺流程大致相同,一般是通过预干燥、预结晶、缩聚和冷却等步骤,预结晶、干燥、固相缩聚可在同一设备中进行,其主体反应器分别为真空转鼓和移动床。间歇式固相缩聚的工艺过程是:预聚体切片首先经筛分、金属分离之后,通过以空气或氮气为加热介质的干燥结晶装置,结晶后进入真空转鼓反应器,抽真空后进行固相缩聚。反应完成后再冷却至160 ℃,再用氮气解除真空状态后出料,或采用程序工艺控制在转鼓反应器中顺序进行干燥结晶、固相缩聚、降温各工艺过程后完成整个反应[45]。反应产生的水和乙二醇等小分子副产物借助抽真空除去,其工艺流程短、操作方便灵活,可以根据所需工业丝性能和用途来调节特性黏度。间歇式工艺是传统的工艺路线的切片传热效率和生产效率低,对真空度要求严格,变换切片灵活性较大,因此适应小批量、多品种生产。
表2 固相缩聚工艺中连续法和间歇法之间的特点比较[47]Table 2 Comparison of characteristics between continuous and batch processes in solid state polycondensation[47] |
| 连续法固相缩聚 | 间歇法固相缩聚 |
|---|---|
| 产物特性黏度较为均匀 | 不同批次产物的特性 |
| 更换原料特性黏度时不够灵活 | 更换原料特性黏度较为灵活 |
| 原料要有较为稳定的来源 | 原料可以有多种来源 |
| 采用氮气除去小分子产物 | 采用抽真空法除去小分子产物 |
| 利用实现自动化,生产效率较高 | 方便把控产品质量,较小的产品CV值 |
| 产能较大,可实现批量生产 | 操作便捷,生产能力小 |
连续式固相缩聚工艺过程大体如下所述:作为预聚体的常规纺丝级PET切片首先经筛分、金属分离后,进入以热空气或氮气作为介质的干燥结晶装置,结晶后的切片进入连续式的固相聚合反应器。物料在立式反应器中借助重力呈活塞状均匀缓慢向下移动,与从反应器下部通入的热氮气流进行热质交换,热氮气流富集从切片内部扩散出的乙二醇、水等缩聚副产物,从反应器上部流出,反应完成的切片从下部送入产品冷凝器再输送至产品贮存罐,或从下部直接送去纺丝[46]。小分子副产物借助惰性气体除去,其工艺流程长,切片传热效率和生产效率高,固相缩聚后的切片特性黏度均匀,变化黏度灵活性好,可以依靠不同的纺丝工艺条件生产不同特性黏度产品。与传统的间歇式相比,连续式固相缩聚因其生产效率高,有利于自动化控制,更适合于大规模现代化生产。国内连续生产工艺的使用大多是引进美国Bepex公司、德国Gemma公司或日本东丽公司等外国公司生产线。
总之,由于反应温度较低(在PET的熔点下),PET树脂虽然在增黏过程中也会出现热降解现象,但是其副作用却相比于熔融缩聚过程大幅降低,使增黏后的PET树脂产品不仅特性黏度显著提升,而且乙醛含量也显著降低;另外,固相缩聚也可以提高PET树脂的结晶度和熔点,使其作为瓶用PET树脂的适用温度范围也随之提高。然而,采用固相缩聚法增黏也有明显的缺点:由于反应温度介于玻璃化转变温度和熔点之间,PET大分子不能移动,只能依靠分子链在无定形区域的移动和反应,所以反应速度很慢,反应时间长,一般需要15~20 h的反应时间;如此低的反应速度会导致低聚酯颗粒彼此结合,不利于排除小分子,使缩合难以继续反应,使产品的分子量及质量受到影响;再有,固相缩聚的步骤相对繁琐、所需工序长、投入设备多,因此生产成本也较高,更适合工业化批量生产,以工业研究为主,很少用于实验室研究。
(3)PET扩链反应挤出增黏技术
扩链反应通常是指:对链端含有活性反应基团的聚合物使用扩链剂或热熔融等手段进行加工,令其在短时间内通过活性链端之间发生的化学反应,实现分子量的显著提升,具体表现为聚合物分子链(聚合度)的长度增长、聚合物特性黏度变大。这个聚合物主链增长的过程就叫扩链,多数扩链反应都会采用扩链剂,但用量很少(一般质量分数在1%左右)。扩链剂通常是一种双官能团化合物,且能够很容易同两种聚合物链末端的活性基团反应,可以加快、有效缩短反应时间,所以也非常适合挤出机上进行反应挤出加工。按照机理,扩链反应可以分为以下四种类型:(1)活性端基聚合物之间的缩合;(2)与低分子偶联剂进行缩合;(3)利用缩聚反应中的链交换反应;(4)活性阴离子链引发的阴离子聚合。
扩链反应在高分子材料合成与改性方面具有非常重要的实际应用价值,例如在PET树脂的回收方面其巨大的应用价值已经显现。当今PET瓶广泛用于饮料、食品、化工、药品包装等领域,引发废旧饮料瓶回收问题,而扩链反应挤出技术是解决PET瓶回收利用问题的有效方法,可以使废旧的PET瓶作为一种新的原料资源,缓解我国PET原料不足的矛盾[5,48]。回收的PET的分子量或特性黏度可以通过扩链反应显著增加,并且分子量的增加也补偿了反应期间的损失。具体是指在对回收的PET树脂进行熔融挤出过程中,添加一定量的链增长剂,使其与PET大分子链的至少两个端基反应以增加PET特性黏度,实现提升分子量的目标。目前,PET扩链剂通常使用可以与—OH或—COOH或同时与两个官能团反应的加成化合物,例如二唑啉、二环衍生物、二酐等[49⇓~51]。然而,一些化合物在反应时都会伴随着副反应,产生副产物,例如碳酸二苯酯(DPC)缩合扩链剂和聚酯反应[52](图3 ),沸点变高,产生挥发性小的分子苯酚,因为苯酚难以除去,会限制反应。因此还有很多问题有待进一步的研究解决。

3.2 PLA树脂的无溶剂生产
3.2.1 PLA简介
PLA是一种典型的具有生物功能、可降解、可再生的高分子材料。因其具有无毒、生物相容性和组织吸收性, 可以完全降解成二氧化碳和水,很早就得到美国食品药品管理局(Food and Drug Administration,FDA)的安全认证,并首先应用到医疗行业[53]。目前,作为绿色环保的降解材料,一般低分子质量的聚乳酸用于药物缓释材料, 而高分子质量的聚乳酸则可以加工成塑料、纤维、薄膜等高分子材料, 用途非常广泛。所以,聚乳酸的合成制备、加工及应用研究最为活跃。人们不但希望PLA本身具有绿色环保的特性,更希望从生产环节就实现PLA的绿色合成、新的制备技术能够进一步降低PLA的生产成本,使其能够像通用塑料那样被广泛应用。
3.2.2 PLA树脂的生产路线
PLA的制备方法主要分为开环聚合法和直接缩聚法两种[54]。PLA的开环聚合法,又称“丙交酯开环聚合法”:第一步是将乳酸在140~180 ℃下通过缩聚制备出低分子量的乳酸低聚物,然后通过200 ℃以上的高温令其发生裂解、脱水环化反应,得到丙交酯粗产物,粗产物再通过重结晶的方式制取高纯度丙交酯;第二步再将上述精制过的高纯度丙交酯通过开环聚合法制备高分子量聚乳酸树脂,其反应方程式如图4 所示,所以也被称作“两步法”。

由于制备的PLA分子量高、产品性能好,所以两步法是目前工业上最普遍采用的聚乳酸的工业化制备方法,如Cargill Dow、 Shimadzu和Dupont等国际化工企业都是使用此方法生产PLA。因此,丙交酯(催化开环)聚合也是迄今为止发展最为成熟、研究历史最为悠久的聚乳酸制备方法。以此法制得的聚乳酸的相对分子量一般在10万以上。若以Sn(Oct)2为催化剂,调控适当的反应条件,仅2~5 h即可获得分子量高达100万的高分子量聚乳酸[55]。除使用恰当的催化剂外,影响丙交酯开环聚合效果的因素还包括:聚合真空度、温度、时间等,但只有丙交酯单体的纯度才是整个聚合反应过程中最关键的因素,只有采用高纯度的丙交酯才能够合成出分子量高、化学结构可控、力学性能较好的PLA树脂产品。但是高纯度的丙交酯需要通过多次重结晶操作才能得到,其过程不但溶剂耗费量大,产品的收率也低。因此,两步法制备PLA虽然在工业生产中比较容易实现,生产出的PLA树脂然分子量高、品质好,但过高的生产成本严重阻碍了聚乳酸的广泛应用。
为降低PLA的制造成本,采用价格相对较低的乳酸单体为基础原料,通过彼此间的相互脱水、酯化,从而逐步缩聚成聚乳酸的“直接缩聚法”(也称作:一步法)得到人们关注,但由于脱水平衡的动力学控制等内在难题,在1995年之前人们还不能通过“直接缩聚法”获得高分子量的PLA[56]。如图5 所示,PLA的“直接聚聚合法”属于逐步聚合机理,通过分析可知:该缩聚反应为可逆反应,且聚合过程中将不断有水分子生成,如果生成的水分不能被及时扩散、排除,就会严重影响聚合反应的正向进行,无法得到高分子量的聚乳酸;有研究表明:如果在反应过程中不进行任何除水操作,所得PLA的分子量只能在l万左右[56,57]。1995年日本三井公司的Ajioka等[58]在PLA的直接缩聚法中使用了大量的高沸点有机溶剂(200~250 ℃):其目的是将乳酸和聚乳酸溶于高沸点的反应介质时,水分易在其中不产生凝聚,反而易形成相分离后被除去,最终制得分子量可高达30万的PLA树脂。这项工作让科研工作者们意识到:改变反应条件、降低体系黏度、增强水分子扩散效果等方法能够有效促进聚乳酸直接缩聚反应的正向进行,从而得到较高分子量的聚乳酸。


3.2.3 PLA树脂的无溶剂生产技术
(1)熔融缩聚制备PLA树脂
乳酸单体通过直接缩聚法制备高分子量聚乳酸树脂的关键在于除水。为推动聚合反应的正向进行,溶液缩聚方式生产聚乳酸时必须使用高沸点的溶剂,通过共沸原理除去缩聚过程中不断产生的水分子和副产物,不断打破反应平衡,推动聚合反应的正向进行。但高沸点溶剂的存在就使得聚合产物的后处理过程变得相对复杂;另外,这些高沸点溶剂往往是含有芳香性的苯环结构,如甲苯、苯甲醚等,在工业化生产中大量使用这类非绿色(或有毒)溶剂,长此以往必然给人类的健康和生活环境带来的严重危害。
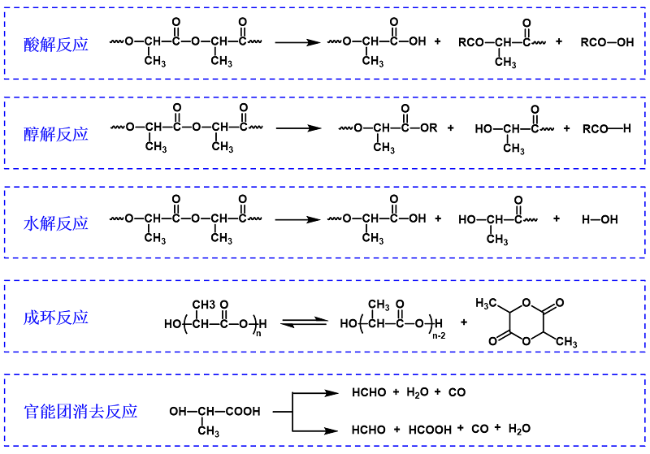
但是,在熔融缩聚的生产过程中,随着反应进行的时间延长,釜内的熔融体系的黏度会越来越大,聚合过程中产生的小分子就越来越难以被排出,导致反应平衡难以向聚合方向移动,尤其是无催化剂下的乳酸熔融缩聚,虽然能够得到高纯度的PLA树脂,但是相对分子质量较低,难以达到实用要求。另外,还有一个严重的问题:从乳酸直接缩聚制备聚乳酸是一个动态平衡反应,在链增长反应过程中还伴随裂解反应和成环反应等副反应的发生,主要包括:酸解反应、醇解反应、水解反应、(丙交酯)成环反应、(官能团)消去反应(图7 )。这些副反应的发生,都是对制备高分子量PLA树脂的严重影响。
所以,在乳酸熔融缩聚法制备PLA的实际生产中存在以下几点问题:(1)熔融缩聚要达到高的分子量必须有足够长的反应时间,一般达20~25 h,但长时间的高温熔融缩聚使得解聚反应严重,因解聚反应生成的丙交酯副产物被不断带出,加速了解聚反应的进行,导致聚乳酸产率偏低,文献报道最高仅60%左右[60];(2)长时间的高温熔融缩聚,容易使聚合物的颜色变深,影响其外观质量和透明性,限制了应用;(3)长时间的高温熔融缩聚,使得聚乳酸的消旋化较为严重,导致聚乳酸的降解速度加快,机械强度降低;(4)最重要的是熔融缩聚得到的聚乳酸,其分子量仍不够高,难以满足应用的要求,尤其是无催化的乳酸熔融缩聚PLA。
为实现采用熔融缩聚法合成高分子量PLA树脂的目的,科研工作者已从动力学控制、水的有效脱除、抑制降解三个关键点着手,开展了大量卓有成效的研究工作。直到2001年,以Moon等[61]为首的日本科研团队才能够在实验室中制备出分子量大于10万的PLA树脂。近20年来,尽管在该研究方向上仍有进一步的突破和发展,但是单纯依靠熔融缩聚工艺制备优质、高分子量的PLA树脂的策略还是不能够完全满足实际生产和应用的要求。为了提高聚乳酸产物的分子量,在熔融缩聚法的基础上又研究了反应挤出、扩链反应和固相聚合等方法。
(2)反应挤出制备PLA树脂
面对熔融聚合技术生产PLA分子量偏低的难题,美国作为PLA树脂的主要生产大国之一,最先提出熔融聚合技术的升级方案——反应挤出技术:以乳酸低聚物或二聚体丙交酯为原料,以双螺杆挤出机为反应器,通过挤出机的挤出模块组合,及对工艺参数的调控,为聚合提供适当的反应环境,在引发剂和催化剂的作用下,聚合单体(丙交酯或低聚物)在通过挤出机进行挤出的过程中,短时间内完成聚合反应,获得高分子量的PLA树脂。
Stevels等[62]研究了采用L-丙交酯在螺杆挤出机中缩聚的方法。当缩聚温度为180 ℃,螺杆挤出机的转速为40 r/min,在挤出机中停留的时间为5 min,催化剂用量为0.5 wt%时,得到了数均分子质量为 6.8万的聚乳酸。1996年,日本JSW公司的Miyoshi等[63,64]报道了反应挤出强化的乳酸熔融缩聚,一步实现了聚乳酸的合成与成型,具体是:将用间歇式搅拌反应器和双螺杆挤出机组合,进行乳酸连续的熔融缩聚实验,结果成功获得了由乳酸通过连续熔融缩聚制得的相对分子质量达15万的聚乳酸。所采用的挤出机经特殊设计,反应料筒被分为5段,各段的温度也可以独立控制,每段都带有1个真空回流装置,一方面可以用来除去水分,另一方面可以同时将反应中生成的丙交醋或L-乳酸低聚物通过回流方式返入料筒中。由于双螺杆挤出机的表面更新速度大大提高,从而使得聚乳酸的分子量也大大提高,最终聚乳酸通过T型头直接制成薄膜制品,一步实现了聚乳酸的聚合和成型。我国开始这项研究比较晚,但发展迅速,并已经达到世界水平。例如,2006年东华大学余木火等[65]发明了另一种采用熔融缩聚法制备高分子质量聚乳酸的方法,是以乳酸、脂肪族二元酸为初始原料,得到了两端都为羧基的乳酸预聚物,然后再加入一定比例的环氧树脂,在一定温度、压力条件下得到了黏均分子质量为 13.0~22.0万的聚乳酸。

总的来说,反应挤出法可一步实现左旋聚乳酸(PLLA)的聚合和成型,具有很好的工业化前景。挤出机的合理设计是其成功的关键,但同时应指出, 该法实现大规模生产可能还有较大的困难,主要在于产量难以提高、设备投资大。
(3)固相聚合制备PLA树脂
因固相聚合温度低,可明显降低因熔融缩聚过程中高温条件下引起的PLA降解副反应,科研工作者也开展了固相缩聚法制备高分子量PLA树脂的研究工作,其反应机理如图9 所示:由于是在低于Tm高于Tg的温度内发生的聚合反应,所以在低分子量的乳酸预聚体(切片、粉末等,主要指通过熔融缩聚制备的PLA)中,大分子链部分被“冻结”形成结晶区,而处于无定形区的分子链末端活性基团、小分子单体和催化剂则可获得足够的能量,通过扩散互相靠近,进而发生有效碰撞,促使聚合反应再次进行;在反应发生的过程中,可以借助真空操作或流动的惰性气体将反应体系中产生的小分子副产物带走(如水),使反应平衡向正向进行,获得更高分子量的PLA。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
当预聚物进行固相缩聚时,高分子量的PLA不断形成,其结晶度也会逐渐升高,此时体系中的小分子物质(催化剂和丙交酯)以及大分子活性端基都聚集在无定形区,可以发生进一步的酯化反应,有利于反应正向进行,促使分子链继续增长,得到较高相对分子质量的产物。这些增长的分子链在晶区与无定形区的边缘聚集,又使得聚合物的结晶度增大。因此,预聚物的结晶度、结晶形态和固相缩聚过程中聚乳酸的结晶度和结晶形态的演变必将对固相缩聚产生很大的影响。
2001年,Moon等[66]在无催化剂条件下制得乳酸的低聚物,然后加入催化剂SnCl2·2H2O(0.4 wt%)和对甲基苯磺酸(TSA,与SnCl2·2H2O等摩尔量),在机械搅拌下加热至180 ℃,并在1 h内将压力逐步减至10 Torr,反应5 h后冷却得到白色固体;将此固体粉碎后在105 ℃的真空条件下预热1~2 h,然后在150 ℃、 0.5 Torr的条件下进行固相缩聚20 h后,得到分子量高达67万的聚乳酸。同时,PLLA的产率达到90%以上,结晶度也得到了明显的提高,且有效地抑制了聚乳酸的消旋。
2002年,汪朝阳等[67,68]以L-乳酸单体为原料,研究了聚乳酸固相聚合的工艺条件,采用熔融聚合得到相对分子质量为5000的预聚物,然后进行固相聚合。结果表明,选用SnCl2作催化剂,60 Pa压力下分段控温反应,聚乳酸的黏均分子量提高到固相聚合反应前的5.3倍(26.5万)。同时研究了固相聚合时间、温度以及分段控温对相对分子质量的影响。同年,宇恒星等[69]通过实验验证了固相聚合方法的有效性,先制得相对分子质量为8000的预聚物,然后以辛酸亚锡为催化剂,得到了黏均相对分子质量为3万的聚合物。通过对固相聚合产物的结晶度和熔点的分析,探讨了固相聚合的机理,由于体系中的催化剂低分子以及大分子端羟基和端羧基聚集在无定形区,发生进一步的酯化反应,向生成聚合物的方向进行,使得分子链进一步增长,得到较高相对分子质量的产物。2004年,钱刚等[70]研究吸水剂应用在聚乳酸固相缩聚反应的可行性,得到了相对分子量达25万的聚乳酸,通过改变催化剂浓度、预聚体粒度和反应时间,研究反应条件对缩聚过程的影响,表明降解副反应仍然是控制聚乳酸最终相对分子量的重要因素。
熔融-固相缩聚可大幅提高聚乳酸的相对分子量,且整个过程为绿色化生产,是一种很有发展前景的制备方法,但此法需降低反应温度、工艺流程长、设备复杂,因此还有广阔的探索空间。
4 总结与展望
从以上的论述中,我们能看到以熔融聚合、反应挤出、固相聚合为代表的无溶剂高分子树脂生产技术是集高分子树脂合成、材料加工制备及工程为一体的新兴科学与技术,是材料科学领域发展的前沿领域,代表着高分子树脂生产技术发展的必然趋势。除采用常规研究方法开展这些研究工作外,我们也希望大家能够注意到在“反应过程中”釜内凝聚态(熔融液态、普弹态、固态)的形成和变化与化学反应的相互影响作用关系,这恰恰是值得我们关注的高分子凝聚态下相关化学问题。在没有溶剂介质的参与下,高分子凝聚态与化学反应之间的相互作用关系变得更加直接、突出,这与我们以往开展的研究工作大不相同,因此在这方面需要开展更多、更细致的基础研究工作,尤其是洞悉高分子凝聚态结构与化学反应之间的联系,从凝聚态化学的角度去发现新问题并找到解决问题的新方法。