



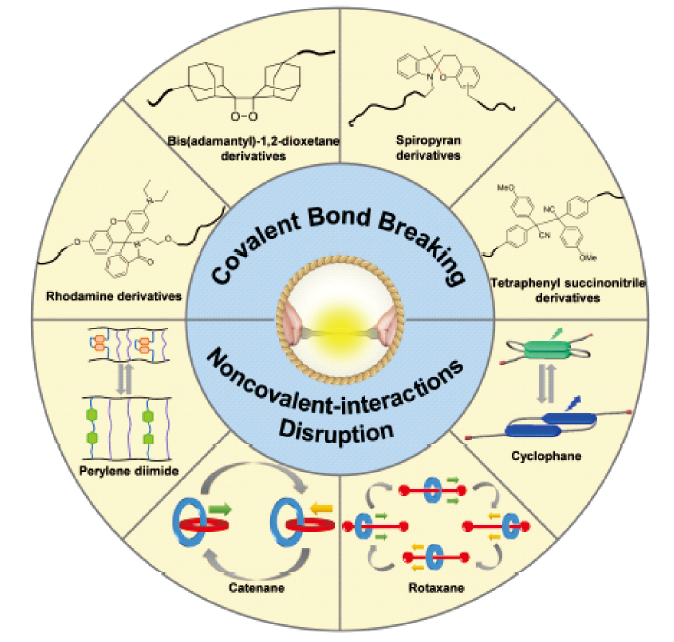

Contents
1 引言
2 利用物理掺杂制备力刺激响应发光聚合物
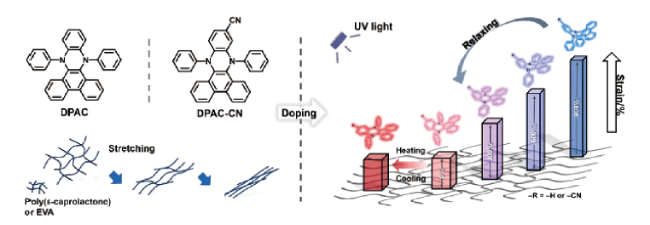
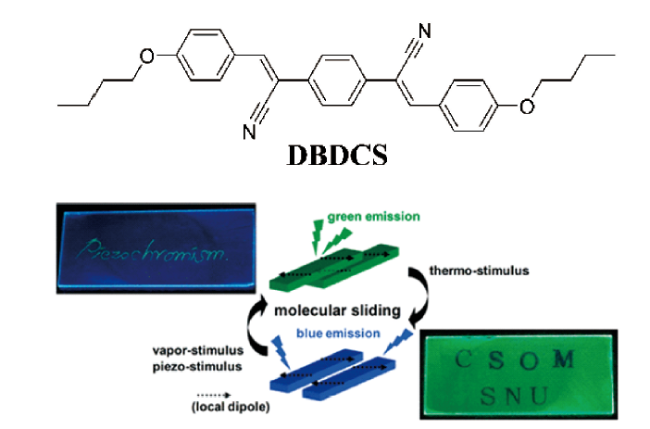
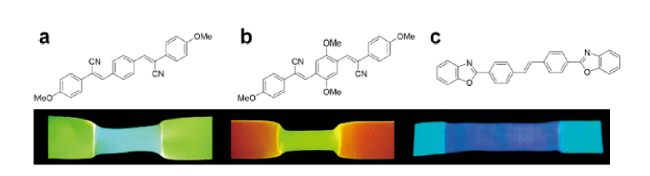
图3 (a) 1,4-双(a-氰基-4-甲氧基苯乙烯)苯;(b) 1,4-双(a-氰基-4-甲氧基苯乙烯基)-2,5-二甲氧基苯[12];(c) 聚丙烯中的4,4'-双(2-苯并口恶唑基)二苯乙烯的分子结构及其对应的聚合物薄膜在拉伸变形后紫外光下的图片[15]Fig. 3 Molecular structures of (a) 1,4-bis(a-cyano-4-methoxystyryl) benzene; (b) 1,4-bis(a-cyano-4-methoxystyryl)-2,5-dimethoxybenzene[12]; Copyright 2003, American Chemical Society; (c) 4,4'-bis(2-benzoxazolyl) stilbene and the images of the corresponding polymer films after tensile deformation recorded under UV light[15]. Copyright 2005, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim |
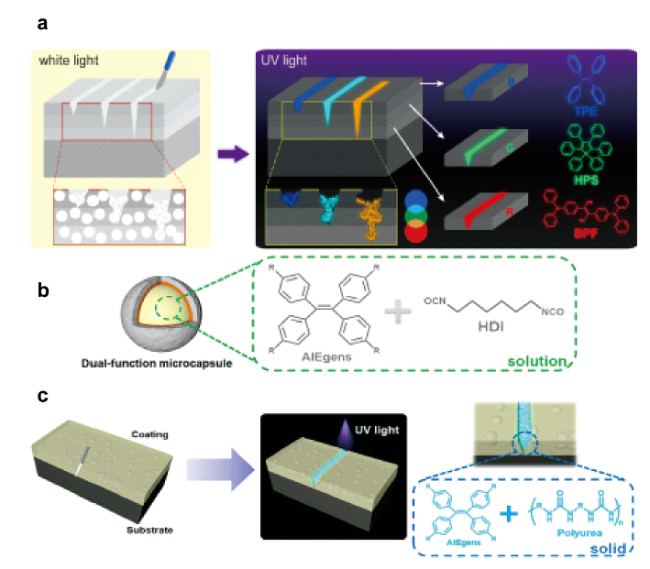
图4 (a) 随裂纹深度变化的多层涂层损伤指示示意图以及具有AIE特性的BPF、HPS和TPE的分子结构[18];(b) 含AIEgens和 HDI溶剂的微胶囊示意图;(c)自主自愈和荧光标记示意图[19]Fig. 4 (a) Schematic illustrations of the damage indication in multilayer coatings with varying crack depth and molecular structures of BPF, HPS, and TPE with AIE characteristic[18]; Copyright 2018, American Chemical Society (b) Schematic illustrations of a one-part microcapsule containing HDI solution of AIEgens; (c) Schematic illustrations of autonomous self-healing and fluorescence labeling[19]. Copyright 2019, American Chemical Society |
3 利用化学键合制备力刺激响应发光聚合物
3.1 基于分子内共价键断裂的力刺激响应发光聚合物
3.1.1 基于螺吡喃衍生物的力刺激响应发光聚合物
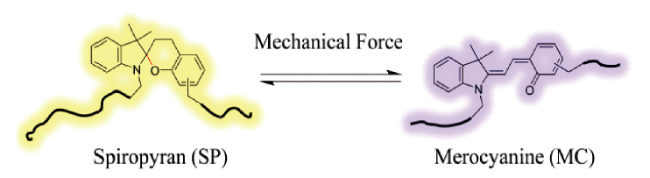
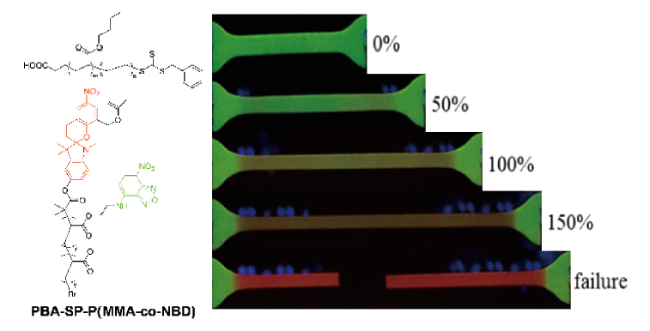
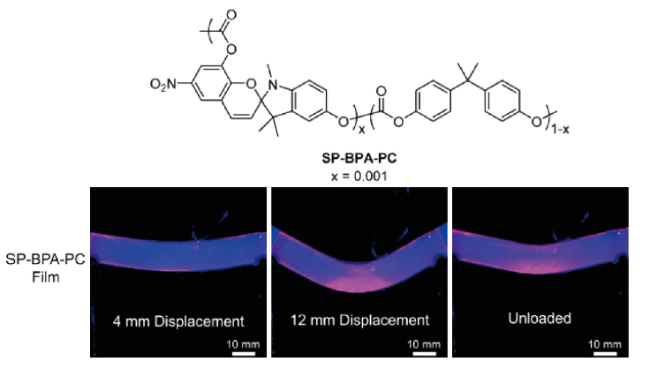
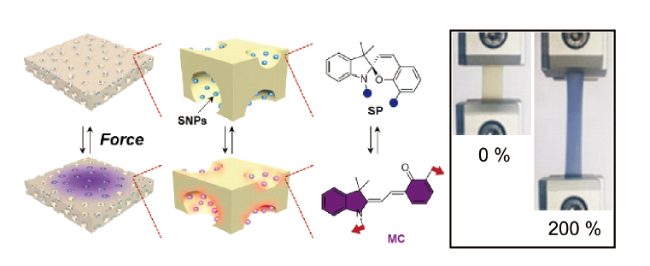
图8 分层NP-MP结构的多孔力致变色复合材料的工作机理示意图和不同结构的力致变色聚合物的颜色变化照片[35]Fig. 8 Schematic of the working mechanism of porous mechanochromic composites with hierarchical NP-MP architecture and photographs of mechanochromic polymers with different structures exhibiting color changes[35]. Copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim |
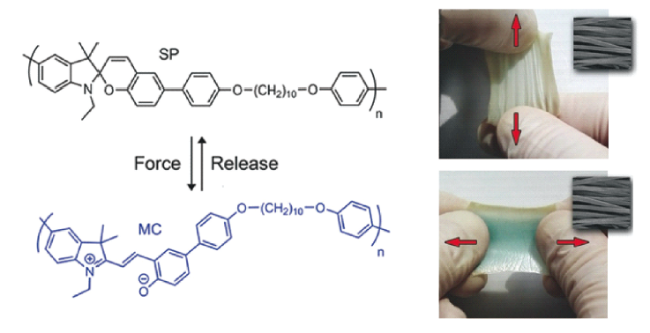
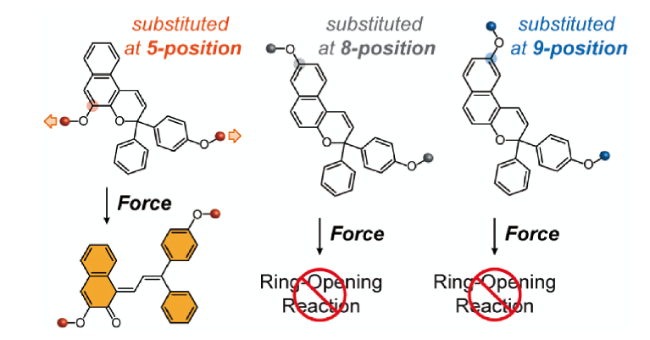
3.1.2 基于双(金刚烷基)-1,2-二氧杂环丁烷衍生物的力刺激响应化学发光聚合物
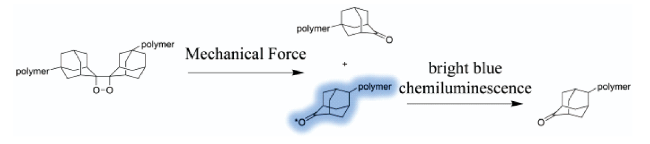
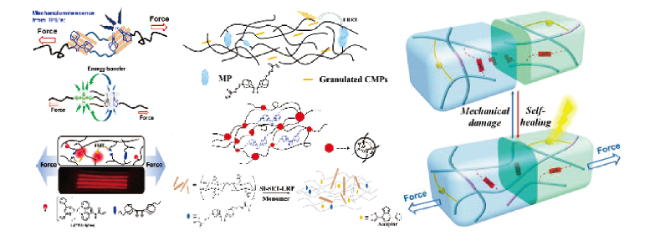
图12 基于双(金刚烷基)-1,2-二氧杂环丁烷衍生物的力刺激响应化学发光聚合物的示意图[42⇓⇓⇓⇓ ~ 47]Fig. 12 Schematic of mechano-responsive chemiluminescent polymers based on bis(adamantyl)-1,2-dioxetane derivatives[42⇓⇓⇓⇓ ~ 47]. Copyright 2018, American Chemical Society; 2014, American Chemical Society; Copyright 2018, American Chemical Society; Copyright 2020, American Chemical Society; Copyright 2019, The Royal Society of Chemistry; Copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim; Copyright 2020, American Chemical Society; Copyright 2020, Chinese Chemical Society |
3.1.3 基于四苯基丁二腈衍生物的力刺激响应发光聚合物

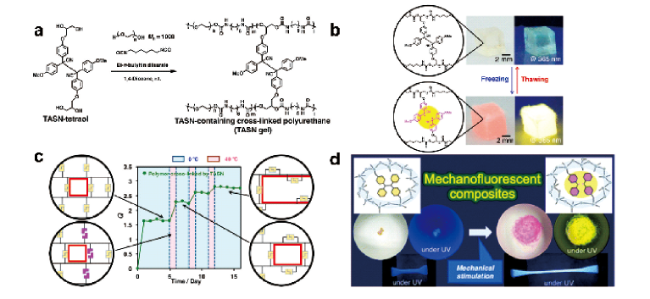
图14 (a) 具有四个羟基的TASN-tetraol的结构及含TASN交联聚氨酯的合成;(b) 环境条件下用紫外光照射(λex= 365nm)经1,4-二氧六环溶胀后的TASN凝胶呈现出冷冻诱导力刺激响变色和冷冻诱导力刺激响应荧光;(c) TASN凝胶网络重组的机理[52];(d) 力激活无色 TASN 与相应的粉红色碳中心自由基之间平衡转换的示意图以及在室温下空气中研磨前后的照片[51]Fig. 14 (a) Structure of TASN-tetraol with four hydroxy groups and synthesis of TASN-containing cross-linked polyurethane; (b) Freezing-induced mechanochromism and freezing-induced mechanofluorescence of TASN gel swollen with 1,4-dioxane under ambient conditions and under UV irradiation (λex= 365nm);(c) Proposed mechanism for the network reorganization in TASN gel[52]. Copyright 2018, American Chemical Society; (d) Schematic illustrations of mechanically triggered conversion of the equilibrium between colourless TASN and the corresponding pink carbon-centred radical and photographs of before and after grinding in air at room temperature[51]. Copyright 2019, Partner Organisations |
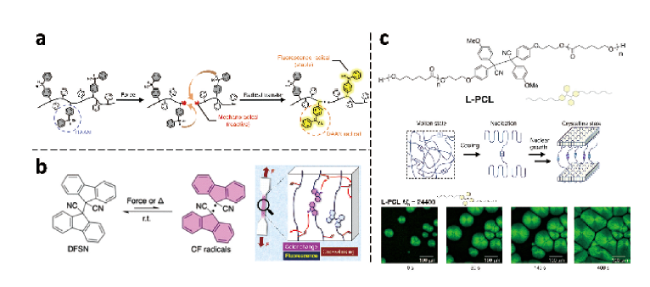
图15 含有(a) DAAN、(b) DFSN和 (c) TASN力敏团的聚合材料的结构和功能示意图[54⇓⇓ ~ 57]Fig. 15 Schematic illustration of the structures and functionalities of the polymers containing (a) DAAN, (b) DFSN and (c) TASN mechanophore[54⇓⇓ ~ 57]. Copyright 2021, American Chemical Society; Copyright 2021, Wiley-VCH GmbH; Copyright 2021, The Author(s) |
3.1.4 基于狄尔斯-阿尔德加合物的力刺激响应发光聚合物

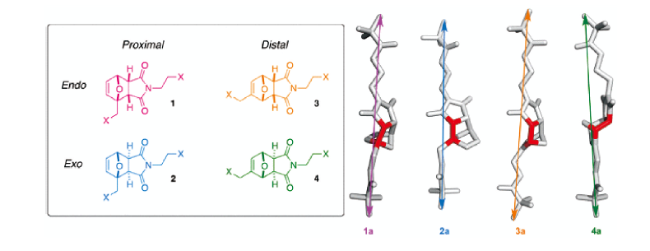
3.1.5 基于梯状烷烃的力刺激响应发光聚合物
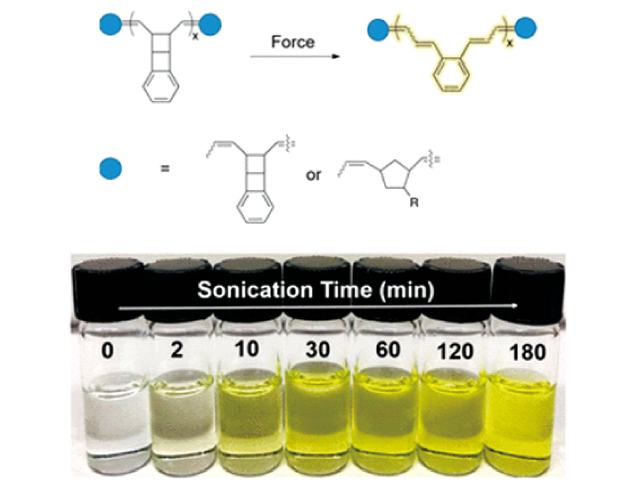
3.1.6 其他基于分子内化学键断裂的力刺激响应发光聚合物
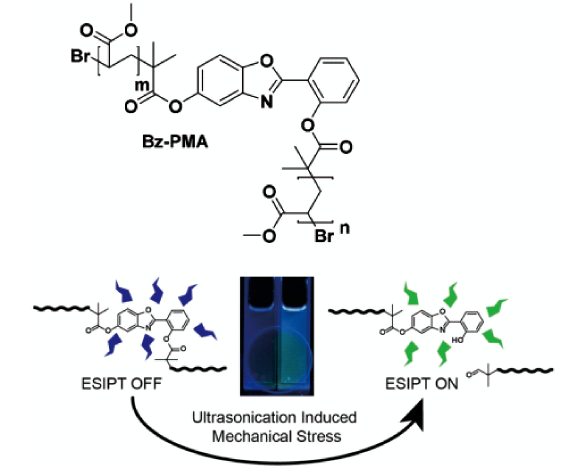
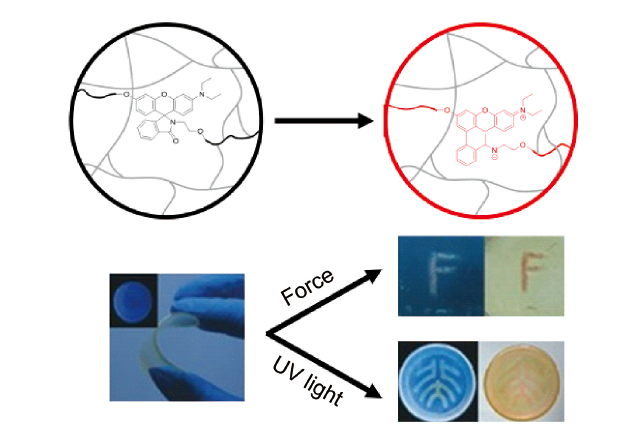
3.2 基于配位键断裂的力刺激响应发光聚合物
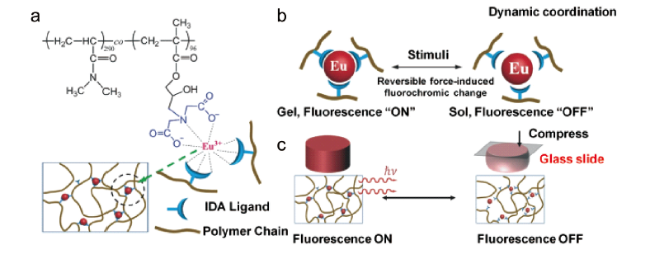
图21 (a) Eu3+离子配位的聚合物水凝胶化学结构示意图;(b) 动态金属配位键驱动的荧光开关和溶胶-凝胶转变示意图;(c) 可逆力刺激响应荧光变色方案[69]Fig. 21 (a) Scheme to show the chemical structures of Eu3+ ions coordinated polymer hydrogels; (b) Schematic illustration of fluorescence turn ON/OFF and sol-gel transition driven by dynamic metal-ligand coordination; (c) Reversible force-induced fluorochromic change[69]. Copyright 2018, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim |
3.3 基于非共价相互作用被破坏的力刺激响应发光聚合物
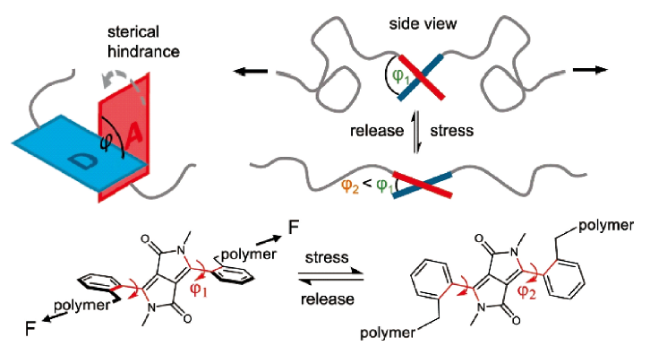
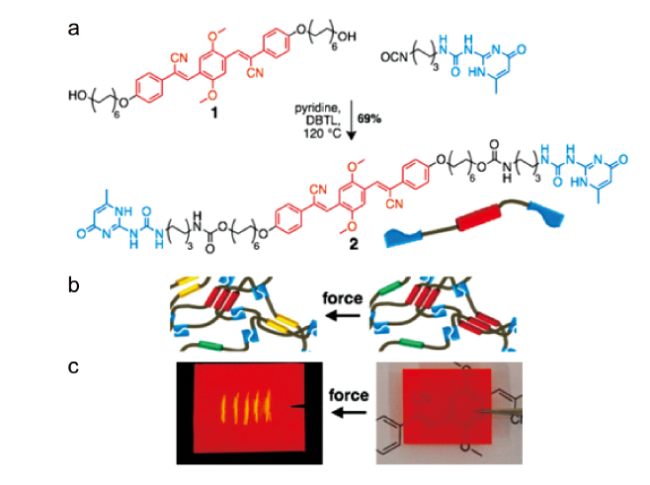
图23 (a) UPy 功能化氰基-OPV 齐聚物UPy-OPV-UPy 2的合成;(b) 超分子组装及力刺激响应发光示意图;(c) 描述力刺激响应发光行为的薄膜图片[77]Fig. 23 (a) Synthesis of the UPy-functionalized cyano-OPV oligomer UPy-OPV-UPy 2; (b) Schematic of supramolecular assemblies and the mechano-responsive luminescence; (c) Images of films, illustrating mechanoluminescent behavior[77]. Copyright 2017, American Chemical Society |

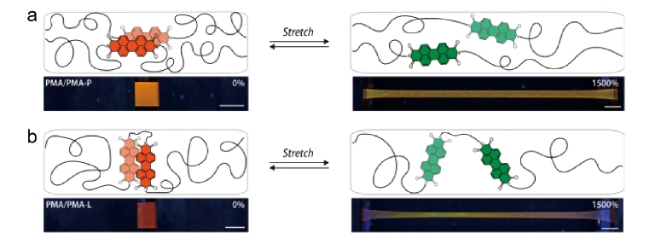
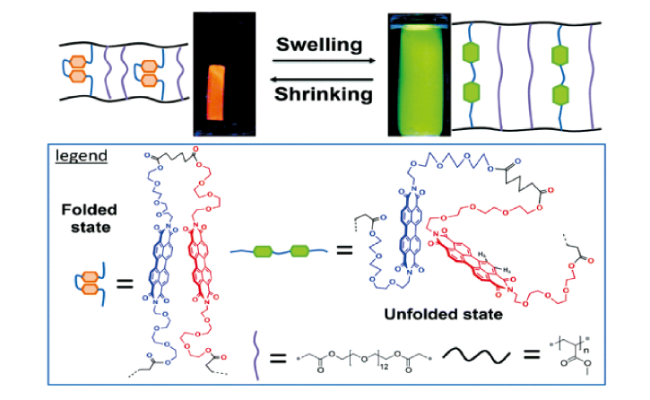

图27 (a)基于环蕃的发光力敏团的分子结构;(b)基于环蕃的超分子力刺激响应发光聚合物结构和力刺激响应发光的示意图[89]Fig. 27 (a) Molecular structures of the cyclophane-based mechanophore; (b) Schematic illustrations of the cyclophane-based supramolecular mechanophore and the mechanochromic luminescence of a polymer containing such mechanophores[89]. Copyright 2021, American Chemical Society |

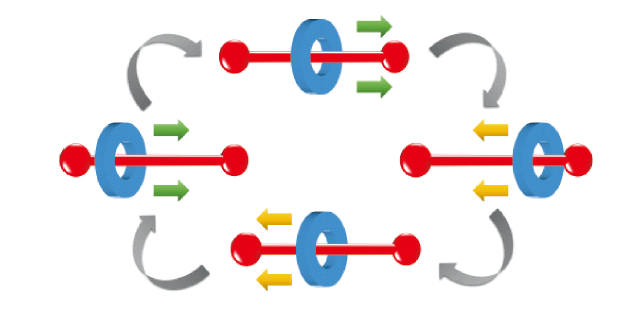
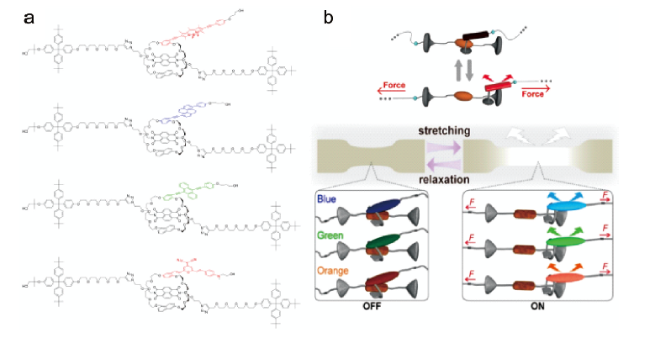
图30 (a) 轮烷化合物的分子结构[26,92];(b) 轮烷体系力刺激响应发光机理示意图[93]Fig. 30 (a) Molecular structures of the cyclic compound[26,92]; Copyright 2018, American Chemical Society; Copyright 2019, American Chemical Society (b) Schematic illustrations of the mechanochromic luminescence mechanism of the cyclic system[93]. Copyright 2019, American Chemical Society |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}