



1 引言
芳香族化合物中芳环C—H键的活化和功能化是学术界长期以来面临的著名难题。通过C—H键的活化,相对廉价的碳氢化合物可以被转化为高附加值的含氧化合物。其中,苯选择性氧化为苯酚是芳环C—H键活化中最具吸引力的反应之一。苯酚是一种用途广泛的重要大宗化工原料,主要用于生产酚醛树脂、双酚A、己内酰胺、苯胺等化学品[1,2]。近年来,苯酚的需求以每年约3%的速度持续增长,到2021年,苯酚的全球总产量有望达到1360 万吨[3]。工业上90%的苯酚产自异丙苯法,通过以下三步生成苯酚:(1)苯烷基化得到异丙苯;(2)异丙苯氧化得到过氧化异丙苯;(3)过氧化异丙苯再分解产生苯酚和丙酮。然而,该生产过程存在诸多问题,例如,能耗高,苯酚收率低(约5%),联产大量丙酮,因产生易爆炸中间物而存在安全隐患,所用的硫酸引起设备腐蚀和环境污染[4⇓~6]。与之相比,用H2O2、O2等环境友好的氧化剂直接氧化苯,一步法羟基化直接制苯酚,是人们期望的绿色工艺,也是长期以来化工领域的研究热点之一[1,7]。然而,苯环的共轭大π键具有很高的化学稳定性,因而苯环上的C—H键活化难度大,导致催化剂的反应活性普遍较低。另外,动力学研究[8,9]指出,苯的氧化为连续的串联反应过程。在反应初期,苯的转化率和苯酚收率均会随反应时间的延长而提高,但生成的苯酚比苯更容易被氧化,苯酚将被继续转化成苯二酚、苯醌,甚至炭黑、焦油等副产物,这是苯羟基化反应的苯酚选择性低、难以提高的主要原因。因此,苯高效定向转化为苯酚仍然是有机合成中最具挑战性的难题之一。
已有一些综述文章总结了不同时期的苯羟基化制备苯酚催化剂的研究进展。例如,2013年,Jiang等[2]综述了以H2O2为氧化剂,掺杂金属的分子筛类催化剂催化苯羟基化的研究进展;2014年,王伟涛等[1]总结了以O2为氧化剂,多种金属基催化剂催化苯羟基化的研究进展;2020年,Ottenbacher等[16]总结了近十年来催化芳香族C—H键氧化的均相及多相催化剂的进展,包含了部分苯羟基化催化剂的内容。近些年,苯羟基化反应的多相催化剂研究取得了诸多重要进展,表征手段的丰富和计算化学的迅速发展,也使研究者们对苯羟基化的催化机理和反应历程进行了更深入的探析。而且,发表的综述文章较少以苯羟基化催化机理作为主要线索进行归纳分析。基于此,本综述主要梳理以环境友好的H2O2/O2为氧化剂,各金属基催化剂催化苯羟基化的反应机理及催化剂构效关系,涉及的文献以近十年发表的为主。特别是对近几年新出现的非金属催化剂催化的苯羟基化反应及机理,也进行了归纳和阐述。本文着重归纳分析苯羟基化的催化机理及相应多相催化剂设计的最新进展,并力求在此基础上加深对该催化反应体系的认识,为进一步研发提高苯羟基化活性和选择性的多相催化剂提供有益借鉴,对推动一步法苯羟基化制苯酚的实际应用进程有所启发。
2 金属基催化剂催化的苯羟基化反应
芳环C—H键选择性氧化有两种途径:(1)通过活化反应底物诱导氧化反应发生;(2)通过活化氧化剂产生活性氧物种进攻反应底物。由于苯环C—H键键能高达473.1 kJ/mol[34],苯羟基化反应很难通过先激活苯环生成苯酚,因此通常以途径(2)为主。苯羟基化反应大多数在温和的液相条件下进行,反应体系由苯(反应底物)、氧化剂(H2O2/O2+还原剂)、催化剂和溶剂组成。本节介绍金属基催化剂催化苯羟基化反应的机理,并讨论催化剂组成结构与催化性能之间的关系。考虑到H2O2和O2作为氧化剂时,它们被催化剂活化的难度和机理都不尽相同,故将二者分别讨论。根据反应过程中产生的活性中间体的类型,以H2O2为氧化剂时涉及自由基机理和非自由基机理;而以O2为氧化剂时涉及自由基机理、非自由基机理以及双活性中心协同催化机理。基于上述不同的反应机理,表1 总结了近年来典型的金属基催化剂催化苯羟基化反应的活性,包括苯酚收率(或苯的转化率)和选择性,以及对应的反应条件。
表1 不同反应机理的苯羟基化反应金属基催化剂的活性对比Table 1 Comparison of activities of metal-based catalysts for hydroxylation of benzene via different reaction mechanism |
| Mechanism | Catalyst | Reaction condition | Activitya | ref |
|---|---|---|---|---|
| H2O2-mediated radical | NaVO3 | C6H6 11.3 mmol, H2O2 19.4 mmol, catalyst 0.2 mmol, 25 ℃, 13 h | Y 13.5%, S 94.0% | 38 |
| [Cu2(μ-OH)(6-hpa)](ClO4)3 | C6H6 60 mmol, H2O2 120 mmol, catalyst 1 μmol,50 ℃, 40 h | X 22.0%, S 95.2% | 12 | |
| Fe(DS)3 | C6H6 11.3 mmol, H2O2 11.3 mmol, catalyst 0.05 mmol, 50 ℃, 6 h | Y 54.0%, S 100% | 40 | |
| [Dmim]2.5PMoV | C6H6 10 mmol, H2O2 30 mmol, catalyst 100 mg, 70 ℃, 4 h | Y 26.5%, S 100% | 25 | |
| [C3CNpy]4HPMoV2 | C6H6 10 mmol, H2O2 30 mmol, catalyst 100 mg, 60 ℃, 2 h | Y 31.4%, S 95.8% | 27 | |
| P-[DVB-VBIM]5PMoV2 | C6H6 10 mmol, H2O2 30 mmol, catalyst 100 mg, 55 ℃, 6 h | Y 23.7%, S 100% | 41 | |
| [VO(acac)2]- grafted PMO | C6H6 4 mmol, H2O2 12 mmol, catalyst 300 mg, 50 ℃, 8 h | X 27.4%, S 100% | 19 | |
| POM@OMP | C6H6 6 mmol, H2O2 18 mmol, catalyst 50 mg, 80 ℃, 10 h | Y 33.0%, S 100% | 42 | |
| Cu2O-rGO | C6H6 1 mmol, H2O2 2 mmol, catalyst 1 mg, 40 ℃, 12 h | Y 21.2%, S 85.5% | 22 | |
| FeOCl | C6H6 10 mmol, H2O2 10 mmol, catalyst 100 mg, 60 ℃, 4 h | Y 43.5%, S 100% | 23 | |
| H2O2-mediated non-radical | [NiⅡ(tepa)]2+ | C6H6 5 μmol, H2O2 2.5 mmol, catalyst 0.5 μmol, 60 ℃, 5 h | Y 21.0%, S 91.3% | 43 |
| [CoⅡ(L3)Cl]Ph4B | C6H6 5 mmol, H2O2 25 mmol, catalyst 5 μmol, 60 ℃, 5 h | Y 29.0%, S 97.0% | 44 | |
| Fe-N4 | C6H6 4.5 mmol, H2O2 55 mmol, catalyst 50 mg, 30 ℃, 24 h | Y 78.4%, S 100% | 45 | |
| V-Si-ZSM-22 | C6H6 5 mmol, H2O2 5 mmol, catalyst 100 mg, 80 ℃, 30 s | Y 30.8%, S> 99% | 13 | |
| O2-mediated radical | VxOy@C-S | C6H6 11.3 mmol, O2 3.0 MPa, catalyst 50 mg, ascorbic acid 0.8 g, 80 ℃, 4 h | Y 9.3%, S 96.0% | 9 |
| V/UiO-66-NH2 | C6H6 11.3 mmol, O2 3.0 MPa, catalyst 50 mg, ascorbic acid 0.7 g, 60 ℃, 21 h | Y 22.0%, S 98.1% | 46 | |
| VOC2O4-N-5 | C6H6 11.3 mmol, O2 1.0 MPa, catalyst 100 mg, 150 ℃, 10 h | X 4.2%, S 96.3% | 47 | |
| O2-mediated non-radical | H7PMo8V4O40 | C6H6 2 mmol, Air 1.5 MPa, catalyst 55 mg, CO 0.5 MPa, 90 ℃, 15 h | Y 28.1%, S 59.3% | 28 |
| POM@MOF@SBA-15 | C6H6 10 mmol, O2 2.0 MPa, catalyst 200 mg,ascorbic acid 0.9 g, 80 ℃, 20 min | Y 6.0%, S> 99% | 48 | |
| PMoV@PCIF-1 | C6H6 22.5 mmol, O2 2.0 MPa, catalyst 300 mg, ascorbic acid 0.8 g, 100 ℃, 10 h | Y 12.0%, S 100% | 49 | |
| PdII(bpym) | C6H6 5.6 mmol, O2 2.0 MPa, catalyst 0.02 mmol,Al(OTf)3 0.04 mmol, 100 ℃, 16 h | Y 3.7%, S 79.7% | 31 | |
| H5PV2Mo10O40 | C6H6 0.5 mmol, O2 1.0 MPa, catalyst 800 mg, 50% H2SO4 5 mL, 170 ℃, 6 h | X 65.0%, S 95.0% | 50 | |
| O2-mediated synergistic catalysis | HMS-HPA(V2)+ Pd(OAc)2 | C6H6 22.5 mmol, O2 2.0 MPa, catalyst 500 mg+10 mg, LiOAc 0.2 g, 120 ℃, 10 h | X 12.2%, S 75.6% | 29 |
| C3N4(580)+PMoV2 | C6H6 45 mmol, O2 2.0 MPa, catalyst 100 mg+400 mg, LiOAc 0.6 g, 130 ℃, 4.5 h | Y 13.6%, S 100% | 26 | |
| g-C3N4+Ch5PMoV2 | C6H6 5 mmol, O2 2.0 MPa, catalyst 12 mg+30 mg,LiOAc 0.06 g, 120 ℃, 4.5 h | Y 10.7%, S> 99% | 51 | |
| SFNC(800)+Ch5PMoV2 | C6H6 5 mmol, O2 2.0 MPa, catalyst 10 mg+30 mg,LiOAc 0.06 g, 120 ℃, 3 h | Y 11.2%, S> 99% | 52 | |
| [DiBimCN]2HPMoV2 @NC580 | C6H6 45 mmol, O2 2.2 MPa, catalyst 550 mg, LiOAc 0.6 g,140 ℃, 17 h | Y 10.5%, S 100% | 53 | |
| FeC(5) | C6H6 45 mmol, O2 2.2 MPa, catalyst 200 mg,LiOAc 0.6 g, 150 ℃, 30 h | Y 14.2%, S 100% | 54 |
a Activity includes Y (or X) and S. Y is yield for phenol, X is conversion of benzene, and S is selectivity for phenol based on the formed phenol plus byproducts. |
2.1 H2O2为氧化剂的自由基反应机理
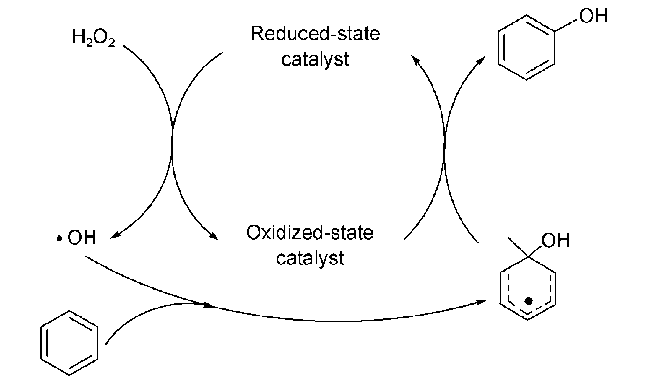
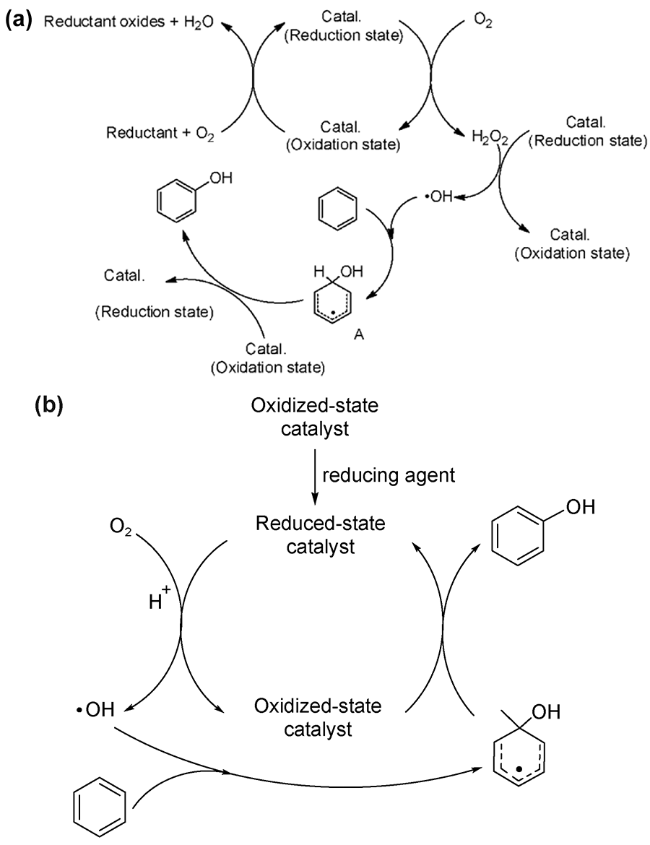
H2O2本身活性较高,在催化剂作用下容易均裂产生羟基自由基·OH[27,35,36]或过氧自由基·OOH[23,37],或与金属键合形成金属过氧自由基M-O-O·[38]。自由基为反应的活性物种,自由基的生成在苯羟基化反应中一般是速率控制步骤[22,39]。反应体系中过渡金属的种类、组成和结构、反应条件(如溶剂种类、溶液的酸碱性等)会影响所产生的自由基类型。图1 给出了一种典型的羟基自由基反应机理。首先,H2O2在催化剂作用下均裂生成活性自由基·OH,进而进攻苯环形成羟基环己基二烯自由基,最终通过氢转移生成产物苯酚。金属基催化剂在反应过程中存在氧化态与还原态的互变循环。早期以Fe2+和H2O2为主要成分的芬顿试剂或者类芬顿型试剂[35,36]催化苯羟基化反应,遵循羟基自由基机理。
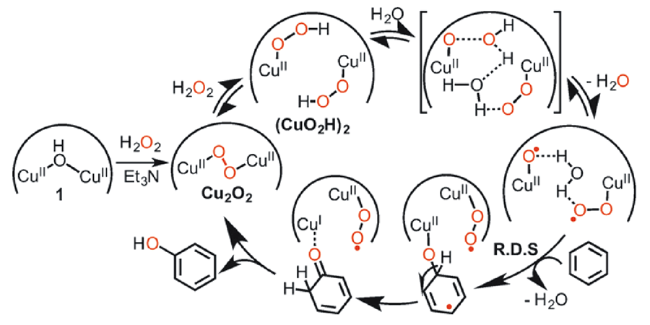
Jian等[38]采用偏钒酸钠作为催化剂,提出反应过程中产生金属过氧自由基M-O-O·机理。反应在酸性介质中进行,钒物种以VO2+的形式存在,随着双氧水加入,VO2+依次形成单过氧钒物种和双过氧钒物种,此后钒氧键均裂生成V(Ⅳ)-O-O·自由基,进攻苯环生成苯酚,完成催化循环。Tsuji等[12]合成了一种均相双核铜分子催化剂,通过计算动力学同位素效应(KIE)值等,提出自由基反应机理(图2 )。催化过程中,在双氧水的作用下,双核铜首先生成具有亲电性质的活性物种前体(CuO2H)2,进一步通过水的氢键作用,(CuO2H)2可逆解离成Cu(Ⅱ)-O·和Cu(Ⅱ)-O-O·自由基,前者进攻苯环生成苯酚。该催化剂效率较高,反应40 h后,苯酚转化数(TON)高达12 000以上。
虽然均相催化剂催化苯生成苯酚的效率较高,但催化剂存在难以回收复用的问题。多年以来,研究者们为提高催化剂的回收复用性进行了持续探索,开发出一系列非均相催化剂以及非均相催化体系用于苯羟基化反应。例如,Peng等[40]将十二烷基磺酸铁作为催化剂,H2O2作为氧化剂,在反应体系中引入离子液体形成非均相催化体系。反应过程遵循芬顿羟基自由基机理,而离子液体介导了相转移过程:催化剂和苯溶于离子液体中,H2O2主要溶于水相中,反应生成的苯酚迅速进入水相,避免了产物深度氧化,提高了反应选择性和转化率。值得指出的是,当反应体系中苯/H2O2和催化剂/苯的摩尔比分别低至1∶1和0.002∶1时,H2O2对苯转化的选择性高达90%。这种液-液两相的非均相催化体系体现出高效、环保的特点,但是催化剂的回收仍是一个难题。
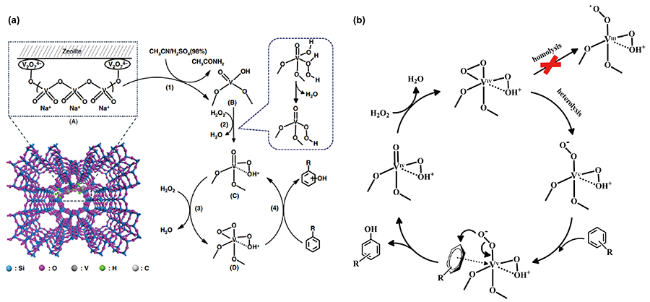
王军等[25,27,41,55⇓⇓~58]采用有机基团或功能化离子液体固化磷钼钒H3+xPMo12-xVxO40的策略,设计得到一类有机离子修饰的杂多酸盐非均相催化剂,如双核离子固体催化剂[Dmin]2.5PMoV2[25]、相转移催化剂[C3CNpy]4HPMoV2[27]、聚离子液体负载型催化剂P-[DVB-VBIM]5PMoV2[41]等,在苯羟基化反应中做了较为系统的研究。通过FT-IR、UV-vis等表征分析,他们认为此类有机离子型杂多酸盐非均相催化剂在反应过程中遵循经典羟基自由基反应机理,H3+xPMo12-xVxO40中的还原态V4+为反应的活性位点。催化剂中的离子液体阳离子与杂多阴离子间存在相互作用,有机组分的修饰不但使得催化剂在反应体系中呈现非均相或相转移的现象,便于催化剂回收,而且加速了V4+/V5+氧化还原可逆循环,提高了磷钼钒阴离子的氧化还原催化性能。
Borah等[19]以CTAB为模板剂,将活性前体乙酰丙酮氧钒与正硅酸乙酯(TEOS)共缩聚合成了负载型催化剂PMO-1,用于苯羟基化反应,发现PMO-1与H2O2形成了V(Ⅴ)-O-O·过氧钒自由基,进而进攻苯环生成苯酚,苯酚选择性高达100%。这是由于PMO载体上的硅醇基团呈弱酸性,弱碱性的苯通过静电作用易被吸附到酸性骨架上,而一旦苯转化为苯酚,酸性的苯酚容易从酸性骨架上解吸,以此防止形成过度氧化产物。Leng等[42]以F127为模板,3-氨基酚和单宁为前驱体,通过煅烧将H5PMo10V2O40封装在有序介孔聚合物(OMP)内,该材料具有窄分布介孔(5~10 nm)和较高的表面积(150~250 m2/g),在催化苯羟基化反应中,显示出较高的催化性能和较稳定的催化剂复用性能。这得益于载体独特的限域效应以及有机组分与PMoV之间的强相互作用。
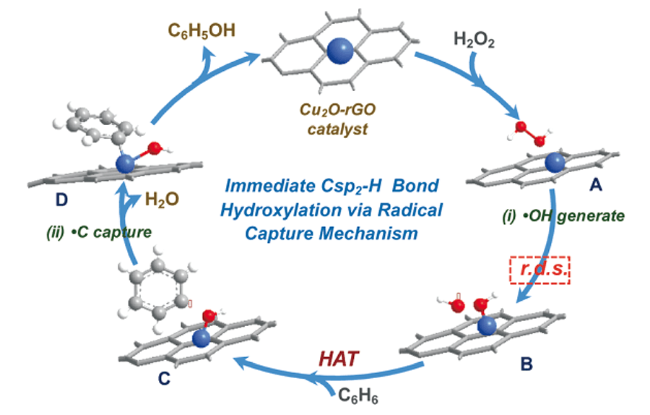
Sun等[22]通过热解金属有机框架(Cu-MOF)与石墨烯,合成了一种非均相复合催化剂Cu2O-rGO,提出了自由基捕获机理(图3 )。首先Cu(I)被H2O2氧化生成Cu(Ⅱ)-OH和·OH,·OH与苯环作用,通过氢原子转移机制(HAT)生成苯环自由基·C,Cu(Ⅱ)-OH能迅速捕获所产生的·C,生成Ar-Cu(Ⅲ)-OH中间体,进而从该高价态Cu(Ⅲ)中间体上还原消除得到苯酚。他们指出,该自由基捕获机制克服了惰性C—H键难以被激活的困难,使得反应更为快速地进行,另外,载体在控制羟基自由基的生成速率、提高反应选择性方面也发挥了极其重要的作用。他们通过对比实验发现,单纯Cu2O NPs分解双氧水速率过快,仅得到6.3%的苯酚收率,而将载体石墨烯rGO与Cu2O NPs复合后,H2O2在Cu2O-rGO上的分解速率适中,同时石墨烯增加了苯的吸附量,两者协同作用,获得了21.2%较高的苯酚收率。
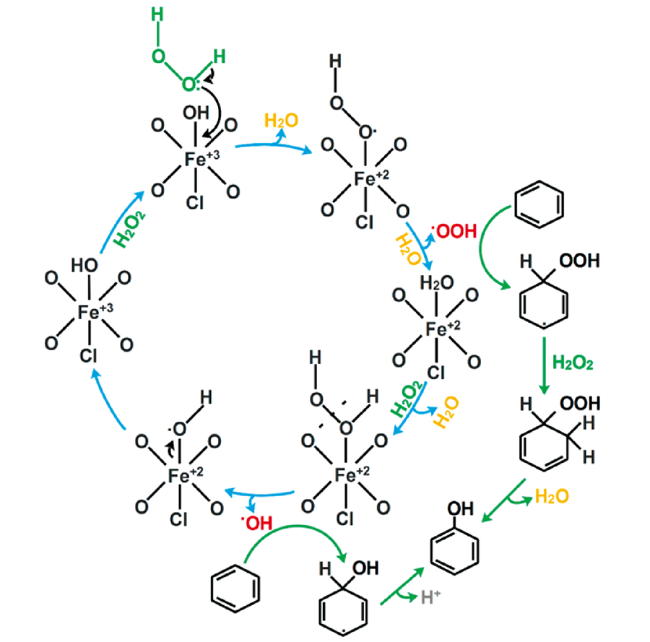
ElMetwally等[23]采用化学气相转换法制备了层状化合物FeOCl,将这种常用的锂电池阴极材料作为非均相催化剂用于催化苯羟基化反应(图4 )。在FeOCl层状框架结构中,铁以Fe3+形式存在,反应过程中的决速步骤(rate-determining step,简称r.d.s)为Fe3+被还原为Fe2+,还原后的Fe2+能吸附H2O2并迅速产生羟基自由基·OH和过氧自由基·OOH,它们分别进攻苯环完成羟基化反应。FeOCl的(020)晶面上分布了大量不饱和Fe原子,以O-Fe-Cl的形式均匀排列,氯、氧原子的配位可以提高铁的自氧化还原能力,得到较高的转化率和选择性。
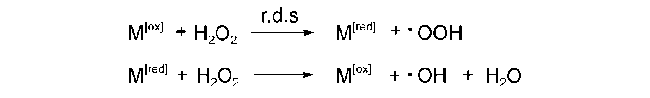
2.2 H2O2为氧化剂的非自由基反应机理
近几年,基于非自由基机理的H2O2苯羟基化反应发展迅速。在非自由基反应机理中,产生的中间体不再是羟基自由基·OH,而是金属-氧活性中间体,如M=O[45]、M-O-M[43]、M-O-O[61]等。KIE值是判断反应属于何种机理的重要手段[13,43]。有报道指出,芬顿型羟基自由基的KIE值一般在1.7~1.8范围内[43,44,62],而金属-氧活性中间体的KIE值往往在0.9~1.1[13,63]。图6 给出了一种典型的苯羟基化的非自由基反应机理,首先H2O2与金属催化剂HO-M-L作用,形成的金属-过氧活性物种  进攻苯环,随后中间体发生氧转移与苯环生成C—O键,最后通过氢转移得到苯酚。一般情况下,氧转移步骤(即C—O键的生成)为非自由基机理的速率决定步骤[45,61]。非自由基机理能促使芳香 —H键的快速活化,并抑制酚类产物的过度氧化,是提高苯羟基化反应活性与选择性的有效方法。
进攻苯环,随后中间体发生氧转移与苯环生成C—O键,最后通过氢转移得到苯酚。一般情况下,氧转移步骤(即C—O键的生成)为非自由基机理的速率决定步骤[45,61]。非自由基机理能促使芳香 —H键的快速活化,并抑制酚类产物的过度氧化,是提高苯羟基化反应活性与选择性的有效方法。
进攻苯环,随后中间体发生氧转移与苯环生成C—O键,最后通过氢转移得到苯酚。一般情况下,氧转移步骤(即C—O键的生成)为非自由基机理的速率决定步骤[45,61]。非自由基机理能促使芳香 —H键的快速活化,并抑制酚类产物的过度氧化,是提高苯羟基化反应活性与选择性的有效方法。
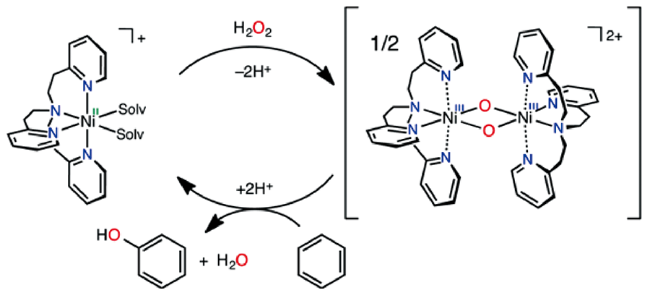
一系列过渡金属均相配合物在苯羟基化反应过程中能够引导非自由基机理。Morimoto等[43]合成了一种镍氮配合物[NiⅡ(tepa)]2+,配体tepa为三[2-(吡啶-2-乙基]胺。在苯羟基化反应中测得的KIE值为1.0,他们推测双镍双氧络合物是反应中间体,该中间体通过亲电进攻芳环发生羟基化反应生成苯酚(图7 )。当使用 O2作为氧化剂时,主要得到18O标记的苯酚,说明双氧水为主要氧源。最近,Anandababu等[44]合成了一种四齿N配体钴配合物[CoⅡ(L3)Cl]Ph4B,反应过程中形成的活性中间体[(L)CoⅢ(OOH)]2+同样通过亲电进攻芳环发生羟基化反应生成苯酚,双氧水同样为主要氧源。这类金属配合物催化反应能够得到21%~29%较高的苯酚收率,但催化剂不易回收复用。
单原子催化剂(Single-atom Catalysts,SACs)凭借其高活性和高选择性的特点,在催化氧化反应领域迅速崛起。为防止单原子团聚,将SACs分散于碳材料的缺陷位上是有效的方法之一。最近,有研究者尝试将此类负载型SACs作为苯羟基化的高效催化剂[64⇓~66]。Pan等[45]通过调控聚合物热解策略,合成具有不同氮原子配位数的单原子铁基催化剂Fe-NxCy。其中,四个氮原子配位的催化剂Fe-N4催化苯羟基化反应得到高达78.4%的苯转化率和100%的苯酚选择性,当Fe-N4的N原子分别被一个或两个C取代成为Fe-N3C和Fe-N2C2时,催化苯羟基化的活性逐渐降低。为了在原子水平上探索结构与反应活性关系,他们对Fe-NxCy SAs/N-C催化体系进行了密度泛函理论(DFT)计算,结果进一步证实了原子级分散的“金属-N-C”结构是催化苯羟基化反应的有效活性位点,不同氮原子配位环境的Fe-NxCy产生的空间结构和电子密度差异是其催化活性改变的主要原因。四配位氮原子锚定的铁基单原子位点Fe-N4活化H2O2的路径所需能垒适中,生成的反应中间体O=Fe=O活性最高,有利于进攻苯环上的C—H键而快速发生氧转移反应(决速步骤),形成稳定C—O键,进而再通过氢转移生成苯酚最终完成催化循环。SACs相对来说结构明确,这给运用计算化学深入认识多相催化机理带来了新契机,该工作的理论计算结果证实,配位模式不但决定了Fe-NxCy催化剂的结构和电子特性,而且影响了反应途径和关键氧化物种的形成,调节SACs的配位环境是提高催化剂催化性能的有效手段,但没有考察该单原子催化剂的复用性能。
综上所述,非自由基机理可以促成苯环上C—H的快速氧化,且相对自由基过程,能避免苯酚发生深度氧化。构筑高苯酚收率和高双氧水利用率的多相催化剂仍是极有挑战性的研究难点,也是实现该反应过程工业化的瓶颈所在。以上以过渡金属催化的双氧水苯羟基化的反应机理为线索,总结了载体、有机基团、配体等催化剂改性手段及效果。值得指出的是,为了提高底物的转化率,多数研究往往需在反应中加入过量的H2O2,这不利于提高H2O2有效利用率,且过渡金属仍然会或多或少地流失在反应体系中,从而影响催化剂的稳定性。
2.3 O2为氧化剂的自由基反应机理
马养民等[9]以生物质(蔗糖、果糖、葡萄糖等)为碳源,NH4VO3为钒源,通过水热法合成了负载型催化剂VxOy@C,在以乙腈为溶剂,反应温度为80℃的条件下,反应10 h后,苯酚收率达到9.3%。通过在反应体系中加入自由基清除剂BHT(2,6-di-tert-butyl-4-methylphenol),证实反应过程遵循羟基自由基机理。体系中的抗坏血酸还原剂协助VxOy@C活化O2产生H2O2,进而生成羟基自由基进攻苯环生成苯酚,但该负载型催化剂的活性位在反应体系中流失严重。为提高催化剂复用性能,他们将VO(acac)2接枝于载体UiO-66-NH2(Zr-MOF)上,制备得到催化剂V/UiO-66-NH2[46],催化苯羟基化反应得到22%的苯酚收率。反应过程中,活性位V4+活化O2产生羟基自由基,带正电荷且具备L酸性的载体UiO-66-NH2有利于反应物苯和羟基自由基的快速吸附,还有利于产物苯酚的及时脱附。此外,将V/UiO-66-NH2进一步煅烧得到的V/ZrO2复用性能较好,苯酚收率也能达到16.2%。
高肖汉等[47]通过水热法合成了氮掺杂的草酸氧钒催化剂VOC2O4-N-5,通过实时原位FT-IR检测,探测到反应过程中生成了超氧自由基[ ·](图10 )。低价态钒V4+首先活化氧气生成高价态钒V5+和超氧自由基[ ·],超氧自由基[ ·]进而与乙酸结合生成过氧乙酸中间体,同时V5+可逆转化为V4+,最终过氧乙酸将活性氧插入到苯环C—H键中生成苯酚。

2.4 O2为氧化剂的非自由基反应机理
 ;O2被活化填充到表面氧缺位生成金属-氧活性中间体
;O2被活化填充到表面氧缺位生成金属-氧活性中间体 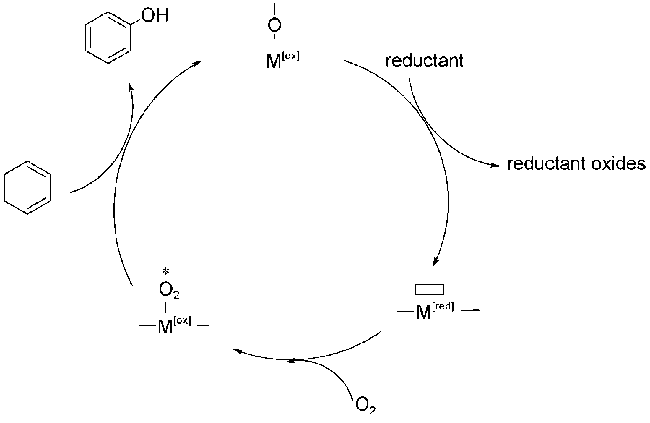
有报道指出[74],钒掺杂的多金属氧酸盐H3+xPMo12-xVxO40中相邻的钒原子(V-O-V结构)容易形成桥式氧缺陷位,这类缺陷位在氧转移反应中具有高活性。Ishii等[28,76]以H7PMo8V4O40为均相催化剂、CO为还原剂进行了苯羟基化反应研究。反应过程中,CO夺取H7PMo8V4O40结构中的氧原子生成氧空位,同时Keggin骨架结构中的V5+被还原为V4+,随后V4+活化O2生成活性氧中间体V5+- ,该中间体进而进攻苯环生成苯酚。通过16O2和 O进行同位素跟踪,证实反应过程中并没有原位生成过氧化氢(H16O18OH)或自由基。产物中少量的PhOH来自H7PMo8V4O40骨架中的氧,反应过程遵循经典Mars-van Krevelen机理。
高爽等[48]设计了金属有机骨架MOF-199包覆的H6[PMo9V3O40]复合结构,并将其负载于介孔材料SBA-15上得到非均相催化剂POM@MOF@SBA-15(PMS)。研究表明,该催化剂在苯羟基化反应过程中组成和结构稳定,仅由于结晶度下降造成PMS重复使用4次后活性稍有降低,反应结束后,在反应体系中未检测到活性组分的流失。
Chen等[49]将PMo10V2 阴离子负载于具有丰富离子结构的多面体低聚倍半硅氧烷(POSS)基多孔离子型框架PCIF-n中,制备得到负载型催化剂PMoV@PCIF-1。该催化材料具有超高比表面积(1025 cm2/g)、超大孔体积(0.9 cm3/g),催化苯羟基化反应获得了较高的催化活性,催化剂复用性能稳定。载体中POSS基的疏水性质有利于亲水性产物苯酚的及时释放,有利于提高苯酚的选择性。
值得关注的是,在上述O2为氧化剂的自由基或非自由基反应机理中,牺牲性还原剂在体系中往往不可或缺,催化剂被还原剂还原为低价态活性位后,才能激活氧气产生中间体促使反应发生。牺牲性还原剂在此过程中发生了不可逆转化,降低了反应的原子经济性并大大增加了成本,不利于反应的实际应用,为此,研究者致力于开发不使用牺牲性还原剂的苯羟基化反应体系[77,78]。例如,Guo等[31]采用成本较高的贵金属配合物PdⅡ(bpym)催化氧化苯环,体系中还引入AlⅢ,通过碱性金属的配位作用有效避免苯环发生偶联副反应,得到较高的苯酚选择性。Neumann等[50]研究发现,在硫酸浓度>50%的水溶液中,苯环的电子被转移到H5PV2Mo10O40结构中,由此被活化产生苯环正离子自由基,进而与O2反应生成苯酚。通过理论计算证实了反应过程遵循电子转移(electron transfer,ET)机制(图12 ),反应需在充足的O2环境下进行。在厌氧条件下,热力学计算结果表明,苯环与POM生成的低自由能中间体不能继续反应生成苯酚。该反应在未添加还原剂的条件下能得到高达65%的苯转化率,但该催化体系的缺点也很明显,例如,催化剂溶于反应介质中不利于回收复用,反应条件苛刻,强酸性条件腐蚀设备且对环境影响也较严重。

2.5 O2为氧化剂的双活性中心协同催化机理
在协同催化领域,双活性位协同催化两个反应底物能够有效降低反应物活化难度,提高反应速率,产生“1+1>2”的效应[53,79⇓~81]。在苯羟基化反应中,从电子转移机制(ET,活化苯环)和晶格氧机理(M-v K,激活氧气)两方面设计构建活化苯环和O2的双活性中心催化剂,有助于在无还原剂参与下快速启动反应并完成催化循环。例如,Liu等[29]将乙酸钯和钒代杂多酸组成协同催化体系,贵金属钯活化苯环变为Pd2+,含钒杂多酸将Pd2+重新氧化为Pd4+,被还原的杂多酸进而活化氧气生成金属-氧活性中间体,进一步进攻被活化的苯环生成苯酚。在不加还原剂的情况下,该体系可得到9.4%的苯酚收率。该反应的主要不足之处是采用成本较高且无法回收的贵金属催化剂。
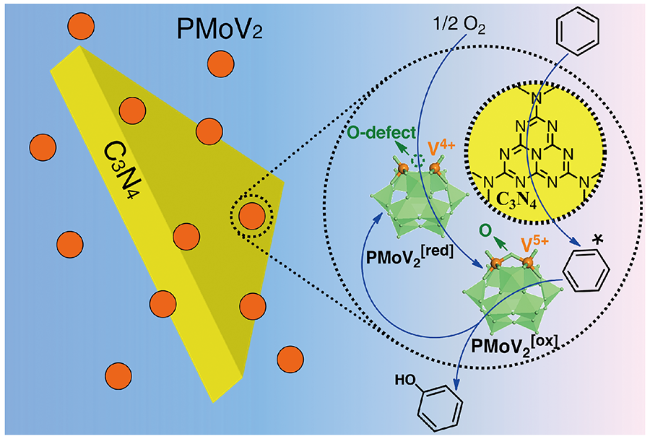
王军等在构建双活性中心协同催化苯羟基化反应方面进行了持续探索[26,51⇓⇓~54]。Long等[26]构建了具有密勒胺结构的g-C3N4和钒代杂多酸PMoV2“混合式”的非贵金属催化体系,提出了双中心协同催化反应机理(图13 ):苯环吸附在g-C3N4上,g-C3N4通过π电子轨道重叠向苯环传递“电子密度”,形成富电子苯环过渡态中间体,同时, 进攻富电子苯活性物种,V-O-V上的桥氧转移到被活化的苯环上形成苯酚,催化剂上随之产生含氧空位的过渡态 ;随后,分子氧氧化 ,插入 的氧空位将PMoV2恢复到起始态 。该催化体系在未添加牺牲性还原剂的条件下,给出了13.6%的苯酚收率。但该催化体系也存在局限性,为了使苯环活性中间体与含活性氧的PMoV2快速接触反应,降低两种反应中间体之间的传质困难,“混合式”催化剂中PMoV2在反应过程中溶于反应体系,有利于与固相g-C3N4的紧密接触,但不方便回收复用。
Cai等[53]将氮掺杂有序介孔碳(NC-580)和杂多酸基有机离子盐([DiBimCN]2HPMoV2)复合,制备得到“一体式”双活性中心非均相催化剂,在最优反应条件下,苯酚收率可达到10.5%,该催化剂通过简单过滤就能回收复用,但难以完全避免杂多酸活性组分从载体表面流失到反应体系中。
Qin等[54]以铁氰化钾为铁源,聚离子液体为碳源,通过高温煅烧得到铁原子高度分散的碳材料FeC。FeC(5)中的类石墨结构碳具有连续的π电子共轭结构,通过π-π相互作用苯环被吸附生成苯活性中间体,随后FeC(5中Fe3+-O-Fe3+的晶格氧原子直接氧化苯活性中间体生成苯酚,而催化剂被还原成含氧缺位的还原态Fe2+-  -Fe2+,最终氧气填补到Fe2+-
-Fe2+,最终氧气填补到Fe2+-  -Fe2+中再生氧缺位。在无还原剂条件下,FeC(5)催化苯环羟基化得到14.2%的苯酚收率,催化剂在反应结束后经过滤即可回收,且复用性能较好。
-Fe2+中再生氧缺位。在无还原剂条件下,FeC(5)催化苯环羟基化得到14.2%的苯酚收率,催化剂在反应结束后经过滤即可回收,且复用性能较好。
-Fe2+,最终氧气填补到Fe2+- -Fe2+中再生氧缺位。在无还原剂条件下,FeC(5)催化苯环羟基化得到14.2%的苯酚收率,催化剂在反应结束后经过滤即可回收,且复用性能较好。以O2为氧化剂的苯羟基化制苯酚是目前原子经济性最高且节能环保的绿色工艺。通过以上对过渡金属基催化剂及反应机理研究进展的梳理可以看出,完成该反应催化循环的难度很大,尤其是在无牺牲性还原剂参与的条件下完成催化循环的难度更大。一方面,对过渡金属活性位点及载体的设计、有机官能团等改性手段的运用、催化剂孔结构及亲疏水性的改善等,将是研究者长期关注的主要研究内容。另一方面,从协同催化机理的角度出发,探索具有双活性中心的非均相催化剂,避免体系中添加牺牲性还原剂,提高催化剂回收的便捷性和复用稳定性,仍是人们追求的目标。
3 非金属材料催化的苯羟基化反应
在金属基催化剂催化的苯羟基化反应中,催化剂中的金属活性组分会或多或少地溶脱在液相反应体系中[9,49,55,77],导致回收复用性能不理想,并引起环境污染等问题。与之相比,非金属催化剂中不含有金属原子或离子,可以避免金属活性组分的流失问题,是一类很有前途的环境友好型非均相催化剂。近几年,以碳催化剂为代表的非金属催化剂已成为苯羟基化反应中最有活力的研究对象之一,探索绿色、高效、循环使用稳定性好的非金属催化剂显示出重要的研究价值。有报道指出[82],以碳材料为代表的非金属材料表面的含氧官能团及缺陷结构,能够作为催化活性位点活化氧化剂,产生自由基活性中间体,从而驱动苯羟基化等多类氧化反应。表2 总结了典型的非金属材料催化苯羟基化的反应活性,从目前已报道的结果来看,无论是以H2O2还是O2为氧化剂,这些催化过程都遵从自由基机理。
表2 非金属材料催化的苯羟基化反应活性对比Table 2 Comparison of activities of non-metal catalysts for hydroxylation of benzene |
| Catalyst | Reaction condition | Activitya | ref |
|---|---|---|---|
| CCG | C6H6 1.67 mmol, H2O2 22.5 mmol, catalyst 20 mg,60 ℃, 16 h | X 18.5%, S 99% | 11 |
| MWCNTs | C6H6 11.3 mmol, H2O2 13.5 mmol, catalyst 50 mg,60 ℃, 2.5 h | X 7.0%, S 97% | 83 |
| CNT7000 | C6H6 11.3 mmol, H2O2 22.5 mmol, catalyst 100 mg, 60 ℃, 36 h | Y 13.7% | 84 |
| HPC-400 | C6H6 130 g, H2O2 22.5 mmol, catalyst 20 mg, 60 ℃, 16 h | X 4.1%, S 99% | 85 |
| WAC-0.5N-8H | C6H6 22.5 mmol, H2O2 58.8 mmol, catalyst 600 mg, 70 ℃, 6 h | Y 13.5%, S 86.3% | 86 |
| NOC-0.15 | C6H6 45 mmol, O2 2.2 MPa, catalyst 200 mg,LiOAc 0.6 g, 150 ℃, 48 h | Y 12.5%, S>99% | 72 |
| AC-70 | C6H6 45 mmol, O2 2.2 MPa, catalyst 200 mg, LiOAc 0.6 g, 155 ℃, 36 h | Y 10.1%, S 100% | 87 |
| QAP | C6H6 5.7 mmol, O2 2.0 MPa, catalyst 100 mg, LiOAc 0.4 g, 120 ℃, 12 h | Y 15.9%, S>99% | 88 |
a Activity includes Y (or X) and S. Y is yield for phenol, X is conversion of benzene, and S is selectivity for phenol based on the formed phenol plus byproducts. |
3.1 碳材料催化苯羟基化反应的自由基机理
碳材料在自然界中广泛存在,具有成本低、无毒、轻质、易功能化、孔隙率可调等优点,是目前研究和应用最为广泛的一类无机非金属材料,主要有石墨烯[89]、碳纳米管[90]、介孔有序碳材料[72]、杂原子掺杂碳材料[91]等。碳材料表面的缺陷位、含氧基团可充当电子给体和受体的角色,在氧化过程中表现出类似于金属的电子得失行为,实现低价态和高价态之间的互变循环[87,92,93]。因此,越来越多的研究者将碳材料作为非金属催化剂催化氧化还原反应[89,94,95]。碳材料的表面积和孔体积[85]、掺杂的杂原子(N、P、B等)[91,96]、缺陷位类型[84](如锯齿型和扶手椅型)、表面含氧基团[72]以及残留微量金属等因素,均有可能对其催化氧化性能产生影响。碳材料催化的苯羟基化反应过程一般遵从自由基机理。例如,石墨碳的sp2杂化表面,存在大π共轭电子体系,可活化氧化剂产生过氧或超氧中间体[97,98];石墨烯等纳米碳材料的边缘或缺陷具有很高的化学反应活性,边缘上未配对电子可活化氧化剂产生超氧中间体[15,85,89];碳材料表面含氧基团,如醌羰基和酚羟基也可作为活性位活化氧化剂,产生羟基自由基活性中间体[86,87,93]。碳材料不但能活化氧化剂(O2或H2O2);而且其大π电子共轭体系能吸附苯环,增加苯环表面的电子密度从而激活苯环[51,52]。由此可见,碳材料在某种意义上也属于双活性中心的非均相催化剂。
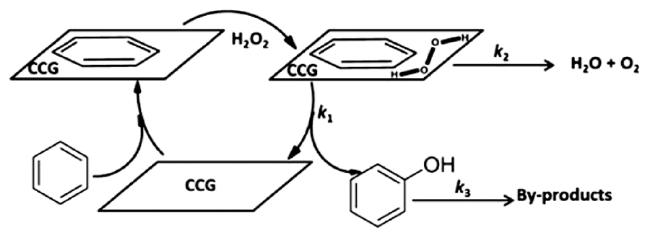
石墨烯纳米碳材料边缘局域增加了额外的电子态,叠加在成键π和反键π*轨道之间,增加了费米能级电子密度,因此石墨烯边缘位点具有活化氧化剂的化学反应活性[89]。同时,石墨烯的大π电子体系具有吸附反应物苯环的能力。马丁等[11]报道了一种经化学处理的石墨烯催化剂CCG,催化苯羟基化得到18%的苯酚收率。在反应过程中,反应物苯和H2O2同时吸附在CCG表面,遵循Langmuir-Hinshelwood机理。吸附在CCG表面的H2O2首先解离为活性中间体,进而与吸附在邻近的苯环反应,以速率k1生成产物苯酚(图14 )。同时,活性氧中间体也会以k2的速率分解为水和氧气。CCG具有平衡k1和k2速率的动力学控制能力,有效避免了苯酚发生深度氧化反应,促使苯高选择性转化为苯酚。
碳材料的缺陷位对催化活性具有重要影响。Song等[83]用浓硝酸溶液和超声处理得到了不同缺陷密度的多壁碳纳米管MWCNTs,将其直接用于催化H2O2为氧化剂的苯羟基化反应。MWCNTs上的缺陷位吸附双氧水产生活性氧中间体,进而进攻苯环生成苯酚。他们发现缺陷位密度越高,MWCNTs分解H2O2生成活性氧的速度越快。Wen等[84]以经过盐酸处理去除金属残留的碳纳米管CNT7000催化H2O2体系下的苯羟基化反应,得到13.6%的苯酚收率。通过小分子模型催化剂对比实验、XPS、拉曼光谱、SIMS、原位红外光谱等表征,证明碳材料表面的缺陷位是反应的活性中心,尤其是扶手椅构型的缺陷位有利于双氧水吸附并产生活性氧中间体,进而进攻吸附在邻近碳表面的苯环分子生成苯酚。Wei等[85]通过H2还原等手段,同样可以增加多孔碳HPC上的缺陷位,他们也发现缺陷位数量越高,HPC的催化活性越高。为了深入认识对于乙腈介质中苯羟基化反应的碳材料表面缺陷位的催化作用本质,Lyu等[15]对此进行了分子模拟计算,结果表明,锯齿构型缺陷位和扶手椅型缺陷位均是产生反应中间体·OH的有效活性位点,且羟基自由基的生成是反应决速步骤。通过进一步的活化畸变模型(activation strain model,ASM)分析,他们认为,锯齿构型的缺陷位对反应过渡态(TS)有更强的稳定化作用,其三重态3z-O的HOMO-LUMO能隙较窄,因而显示出更高的催化性能。
碳材料表面被浓HNO3、过氧化氢等处理后,表面生成的大量含氧基团也被证明是氧化反应中的催化活性位。报道指出[99,100],碳-氧双键与金属-氧双键具有相似性,碳材料表面的醌羰基和酚羟基发生可逆转化的过程中存在电子得失现象,与金属-氧中间体类似具有氧化还原性能,能够催化氧化还原反应[92,101]。Xu等[86]采用硝酸和双氧水氧化、高温热处理等方法对商用木质活性炭进行改性处理,这种改性活性炭催化的H2O2为氧化剂的苯羟基化反应,取得了13.5%的苯酚收率,催化剂重复使用性能稳定。表征及实验证实,活性炭表面酚羟基与醌羰基之间的可逆循环促使H2O2被活化生成羟基自由基,进而进攻苯环生成苯酚(图15 )。他们观察到,溶液中适当的H+浓度有利于醌/酚建立互变异构平衡。

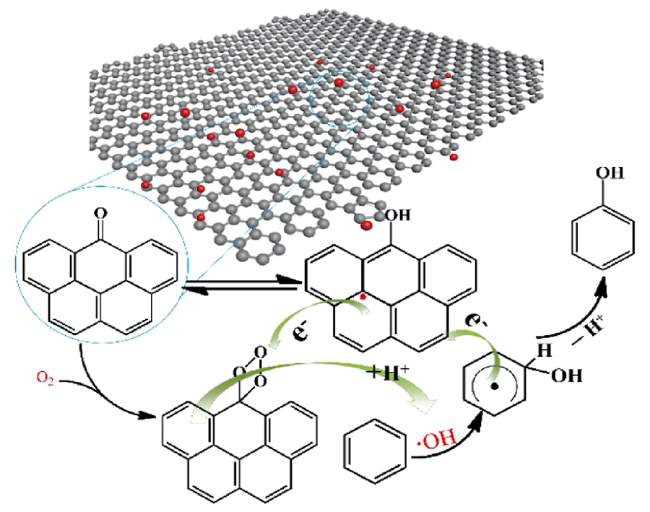
王军等[72,87]对碳材料的表面官能团改性及其在O2作为氧化剂的苯羟基化反应中的催化作用机制进行了研究。Shan等[72]以三聚氰胺改性的酚醛树脂为原料,采用高温炭化法合成了表面富氧的N掺杂介孔碳材料NOCs,表面积达到620 m2/g。在O2作为氧化剂,添加乙酸锂助剂(LiOAc)的情况下,NOCs能高效地催化苯羟基化生成苯酚,苯酚收率高达12.5%,该收率与部分过渡金属基催化剂的结果相当。催化剂重复使用6次后性能仍然稳定,苯酚收率基本不变。经表征、对比试验和DFT计算表明,NOCs表面羰基(环酮类/醌羰基C=O)首先通过化学吸附O2延长O—O键,随后在体系H+的辅助下O2被激活生成·OH,进而进攻苯环生成苯酚(图16 )。他们指出,醌/酚基的互变异构为电子传递提供了前提,氮掺杂不仅提高了NOCs的稳定性,还增加了NOCs表面的醌/酚基数量。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.2 高聚物催化苯羟基化反应的自由基机理
2019年,Wang等[88]合成了一种非金属醌胺类聚合物QAP,该催化剂在催化苯羟基化反应过程中同样存在醌/酚之间的互变循环。在无还原剂的条件下,添加LiOAc作为助剂,QAP催化苯与O2反应生成苯酚的收率高达15.9%。催化过程中O2首先被酚羟基活化产生H2O2,进而均裂成·OH自由基进攻苯环形成羟基环己基二烯自由基,同时酚羟基被可逆氧化生成醌羰基,醌羰基促使羟基环己基二烯自由基发生氢转移生成苯酚。QAP中存在的π-π共轭体系,能够通过协同吸附苯环来提高苯羟基化反应的活性。但是,QAP催化剂重复使用两次后,苯酚收率大幅度降为6.6%。
4 结论与展望
采用H2O2/O2作为绿色氧化剂定向氧化苯环的$\text C_{\text s \text p^{2}}— \text H $键生成苯酚的直接羟基化反应,极具应用前景,但至今仍极具挑战性。针对该反应中苯环上$\text C_{\text s \text p^{2}}— \text H $键难以被活化、氧化剂特别是分子氧的活化难度大、产物苯酚易被过度氧化、催化剂复用稳定性难以提高等多方面的难题,构筑高活性、高选择性、高稳定性的非均相催化剂是主要研究内容,也是推动苯羟基化反应工业化应用的关键。而对催化反应机理及催化剂构效关系的深入认识,是理性设计和构筑高效催化剂的前提。
无论对于金属基还是非金属催化剂,通过活化氧化剂(H2O2/O2+还原剂)而引导的经典自由基机理一直是研究的重点内容,也是人们探索新催化剂时的主要依据。例如,对非金属催化剂,人们近来同样观察到了反应过程中自由基的存在,初步证明表面缺陷位或含氧官能团是导致自由基产生的活性位点。自由基中间体难以避免对目标产物苯酚的进一步氧化,因而不利于提高苯酚收率和选择性。相比之下,非自由基机理具有提高苯酚选择性的潜力。金属基催化剂与H2O2相互作用产生的金属-氧中间体可经亲电甚至亲核氧化路径完成反应;对于分子氧介导的反应,金属基催化剂的晶格氧通过形成氧缺陷位,遵循Mars-van Krevelen机理完成催化循环。为了避免牺牲性还原剂的加入,能够同时活化O2和苯环的双活性中心催化剂及相应的协同催化机理受到重视,并取得可喜进展。在催化机理研究方面,结合原位表征技术的反应中间体的捕获和分析,以及借助计算化学对反应过渡态的剖析,将大大加深人们对苯羟基化机理的认识。与此同时,催化剂活性位点的理性设计和精准制备、载体的选用及其与活性位点的协同作用、有机官能团等助催化剂的修饰、催化剂孔结构构筑及亲疏水性的改善等,仍将是人们长期研究的重点。在追求高效催化性能时,还应特别关注双氧水有效利用率不高的问题,以及如何在温和条件下提高O2为氧化剂时的反应效果难题。
附表 催化剂缩写的英文全称说明Supplementary Table Abbreviation for catalyst and corresponding English full name |
| Abbreviation for catalysta | English full name |
|---|---|
| AC | activated carbon |
| bpym | 2,2'-bipyrimidine |
| C3CNpy | N-butyronitrile pyridine |
| CCG | chemically converted graphene |
| CNT | carbon nanotube |
| Dmim | 1,1-(butane-1,4-diyl)-bis(3- methylimidazolium) |
| DiBimCN | 4,4'-butyl-bis(3-cyanopropyl-imidazole) |
| Fe(DS)3 | Ferric tri(dodecane sulfonate) |
| 6-hpa | 1,2-bis[2-[bis(2-pyridylmethyl)aminomethyl] -6-pyridyl]ethane |
| HMS | hexagaonal mesoporous silica |
| HPA | heteropolyacid |
| HPC | honeycomb-like porous carbon |
| L3 | N1,N1-bis((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)-N3,N3-dimethylpropane-1, 3-diamine |
| MOF | metal organic framework |
| MWCNTs | multi-walled carbon nanotubes |
| NOC | N-doped and surface O-enriched mesoporous carbons |
| OMP | ordered mesoporous polymer |
| P-DVB-VBIM | poly divinylbenzene- 3-n-butyl- 1-vinylimidazolium |
| PMoV | H3+xPMo12-xVxO40 |
| PMO | periodic mesoporous organosilica |
| POM | polyoxometalate |
| PCIF | porous cationic framework |
| QAP | quinone-amine polymer |
| rGO | reduced graphene oxide |
| SNFC | soybean flour prepared nitrogen-doped carbon |
| tepa | tris[2-(pyridin-2-yl)ethyl]amine |
| V-Si-ZSM | V-containing all-silica zeolite |
| WAC | wood-based activated carbon |
a With an alphabetical order of the first letter in abbreviations. |