




1 引言
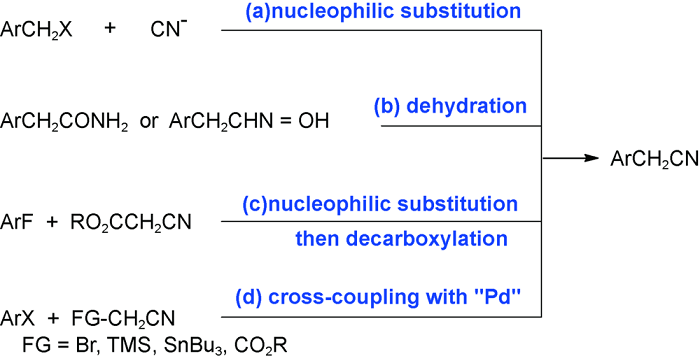
芳基乙腈衍生物是一类重要的中间体,它可以作为合成胺类、羧酸和含杂环二芳酮类衍生物的原料[4,5]。因此芳基乙腈衍生物在药物合成、颜料、染料以及杂环的合成领域得到了广泛应用[6]。制备芳基(杂环芳基)乙腈衍生物的方法主要有通过苄基卤代物与氰化物发生亲核取代反应(a)[7],通过酰胺或醛肟化合物的脱水反应(b)[8],通过芳基氟与氰乙酸乙酯发生取代反应进而脱羧(c)[9],以及Pd催化偶联芳基卤代物与官能化的乙腈衍生物(d),如溴乙腈、TMS-乙腈、Bu3Sn-乙腈、氰乙酸盐等[10⇓~12]。尽管这些方法得到广泛的应用,但上述方法仍具有许多缺点,包括:(1)过渡金属催化剂的价格昂贵,并且可能需要配体和各种添加剂;(2)反应条件苛刻(例如高温或较长的反应时间);(3)需要预官能化的偶联底物[13]。此外,这些反应还会产生对环境和生物有害的有毒重金属废物以及较差的原子经济性。因此,开发出实用、易于操作、低成本、无需过渡金属催化且能够区域选择性地直接形成C—C键的合成方法是非常有必要的。
2 乙腈对活化烯烃的自由基氰甲基化
一般来说,由于氰基的吸电子作用以及可以稳定自由基中间体,因此,切断乙腈分子中α-C—H键于其他类型的sp3 C—H键容易发生,且生成的α氰碳自由基可以容易参与各种自由基反应。
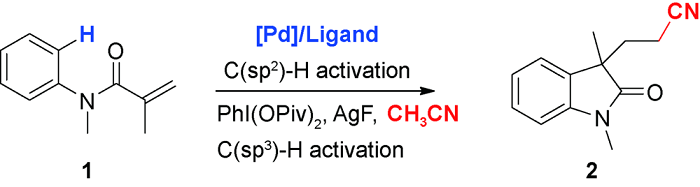
对烯烃的较有效直接氰甲基化反应,最早是2011年Liu等[16]以N-芳基丙烯酰胺(1)为原料,报道了通过一个Pd催化且配体参与的C(sp2)-H 以及C(sp3)-H串联激活的方法与乙腈分子直接组建氰基取代的吲哚酮衍生物(2),该反应用到了金属Pd试剂及含N配体,并且需要适量的氧化剂PhI(OPiv)2以及必需的AgF催化。反应条件比较苛刻且底物适用性范围较少。
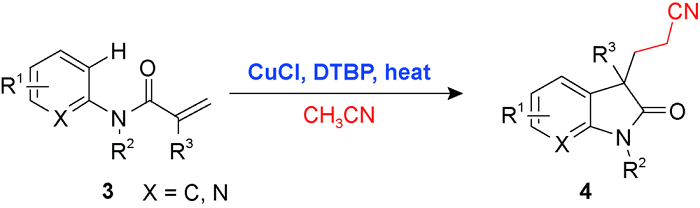
2014年,You等[17]报道了一类活化烯烃的自由基串联氰甲基化反应制备氰基取代的氧化吲哚衍生物。该反应在DTBP自由基引发剂存在下,产生活化氰甲基自由基,进而对烯烃底物(3)进行加成和环化反应,可以高效生成吲哚酮衍生物(4)。该反应具有操作简单、原料简单易得且底物范围广等优点。他们考察了苯环以及杂环吡啶等底物拓展,并且首次制备了硝基取代的吲哚酮衍生物。
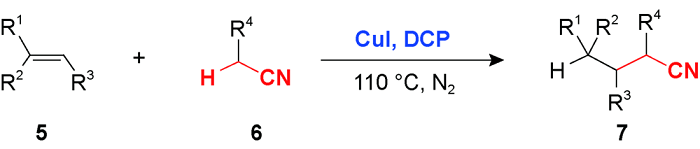
Liu等[18]2015年报道了一类自由基引发选择性激活芳基乙腈(6)中的α-C(sp3)—H键,进而与未激活的底物双键(5)发生反Markovikov加成制备一系列乙腈衍生物(7)。经过条件筛选发现,CuI与DCP引发剂作用下,可以高效地完成该反应的转化,制备各种取代的腈衍生物。该方法具有适用于简单的烯烃,原料简单,且操作温和、适合工业放大等优点。
2015年,Zhu等[19]报道利用Fe促进的自由基氰甲基化反应通过N-芳基丙烯酰胺(8)与乙腈直接制备吲哚类衍生物(9)。该反应涉及一步C(sp3)—H及C(sp2)—H双C—H键官能团化。该反应对甲氧基、乙氧基酰基、氯、溴、碘、硝基、三氟甲氧基以及三氟甲基等具有很好的官能团兼容性,且反应条件温和。
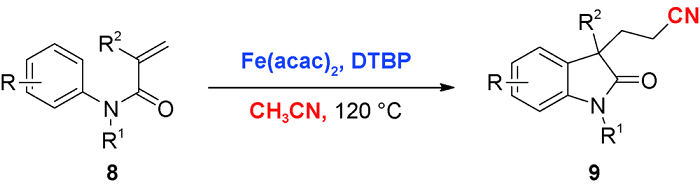
2015年,Zhu等[20]报道烯丙基醇(10)与非活化的烷基乙腈衍生物(11)在铜催化下的烷基化及环氧化制备官能化的环氧化合物(12)。自由基引发剂诱导产生烷基氰自由基,进而对双键进行加成,借助铜辅助催化形成C(sp3)—O键进而形成环氧化合物。该反应提供了一条有效的通过形成C(sp3)—C(sp3)以及C(sp3)—O 键制备双官能团化的三或四取代的环氧化合物的方法。
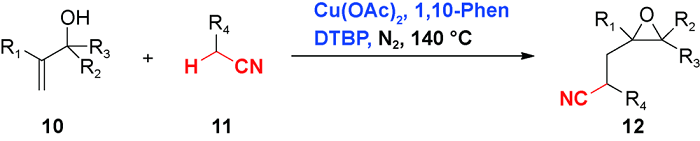
2015年,Zhu等[21]报道Cu-催化氰甲基化烯丙基醇衍生物(13)通过伴随1,2-芳基迁移高效合成含有α-季碳中心的官能化的酮衍生物(15)。反应机理涉及到自由基引发,以及酮催化伴随的neophyl重排,该反应也提供了一条制备含酮以及氰乙腈双官能团衍生物的方法。
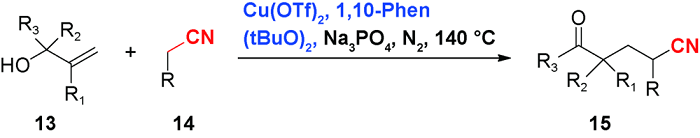
2017年,Li等[22]报道一种自由基参与的氧化分子内1,2-烷胺基化,通过烷基乙腈衍生物(17)、胺以及烯烃衍生物(16)三组分一步构建γ-氨基烷基氰衍生物(18)的方法,该反应利用简单的碳酸银以及铁路易斯盐作为催化剂,高效、简洁,一步构建含C-C、C-N键的乙腈衍生物。
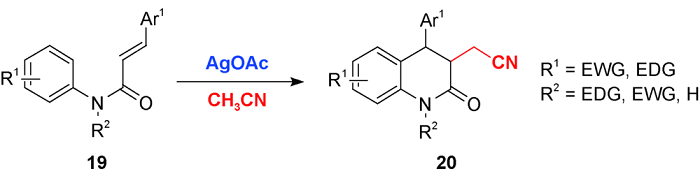
Zhang等[23]2018年报道了一类AgOAc促进自由基氰甲基化反应制备喹啉酮衍生物(20),该反应借助引发剂产生氰甲基自由基,对底物(19)中双键加成伴随分子内环化芳香化反应,进而氧化制备了一系列新颖的3,4-二氢喹啉酮衍生物(20)。该反应操作简单、有很好的官能团兼容性,且适合放大制备克级产品。
3 乙腈对烯烃或芳香C(sp2)—H官能化的光化学偶联反应
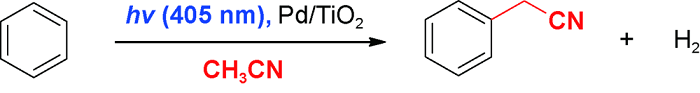
Yoshida等[24]2013年报道了一类钯和二氧化钛催化剂联合使用对苯环进行氰甲基化反应,二氧化钛活化乙腈形成氰基甲基自由基,钯催化剂促进连续取代形成C—C键。该反应在温和条件下进行,减少了有害离去基团的处理,体现了绿色和可持续化学理念。
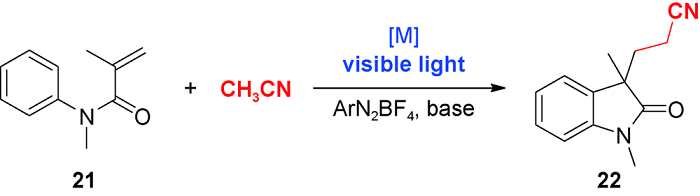
2014年,Li等[25]报道一种光促进对激活烯烃双官能团化策略合成系列吲哚酮衍生物。当配体[Ru(bpy)3Cl2]、自由基引发剂ArN2BF4以及碱存在下,通过36 W荧光灯照射,各种N-芳基丙烯酰胺(21)与乙腈或丙酮发生1,2-烷基芳基化,高收率制备系列吲哚酮衍生物(22)。
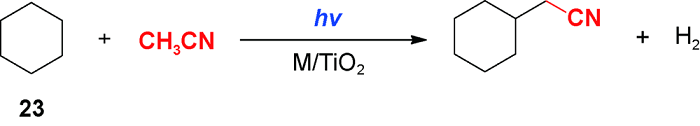
2017年,Yoshida等[26]报道利用(Pt/TiO2)光催化剂直接催化氰甲基化脂肪环或芳环C—H(23)与乙腈发生氰甲基化反应。相对于(Pd/TiO2)光催化剂,(Pt/TiO2)光催化剂被发现在催化脂肪环C—H氰甲基化方面更有效,而催化芳基C—H氰甲基化,则需要金属Pd的参与。在这个过程中,金属Pt和Pd纳米颗粒负载的二氧化钛光催化剂可以作为光激发电子的受体来增强光催化反应速率。
4 乙腈对芳环C(sp2)—H官能化的脱氢偶联反应
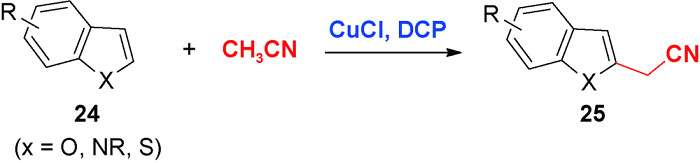
Li等[27]2016年报道了一类新颖的自由基引发特定位点偶联脱氢反应制备杂环芳乙腈衍生物。该反应的底物可以是呋喃、吡咯、噻吩等杂环分子(24),他们探讨了自由基引发剂、一价铜衍生物等,最终发现在DCP及CuCl作用下可以高收率地制备杂环芳乙腈衍生物(25),首次将芳乙腈化发展到杂环分子上。
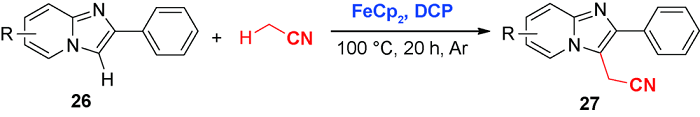
Xu等[28]2017年报道一类2-芳基咪唑并吡啶(26)与乙腈在铁催化下脱氢sp3-sp2偶联反应,该反应在DCP自由基引发剂下,二价铁配体催化下,可以高效直接地对sp2 C—H脱氢氧化制备杂环芳乙腈衍生物(27),且具有底物拓展范围广、收率高、条件温和等优点。
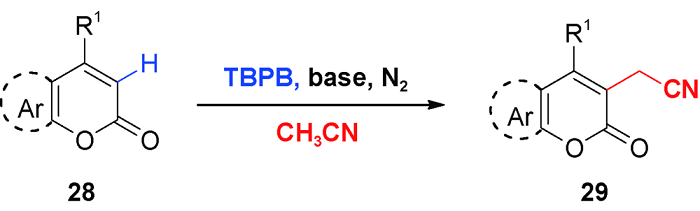
Yan等[29]2018年报道了一类香豆素类化合物(28)在TBPB和氟化钾存在下与乙腈通过直接氧化激活C(sp3)—H的非金属催化的交叉脱氢偶联反应,制备一系列香豆素乙腈衍生物(29)。该反应有良好的官能团耐受性,以及较好的收率,提出了一种自由基途径进行的乙腈氧化C—H活化的新策略。
5 导向基团促进的C—H氰甲基化
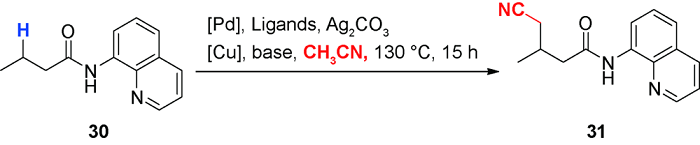
2016年,Ge等[30]报道一种Pd催化配体促进的位点选择性的使用未激活的C(sp3)—H键(30)和乙腈直接发生氰甲基化反应。该反应机理涉及金属Pd与二齿型氨基喹啉配位,碱性条件下远端激活C(sp3)—H键形成环金属二价Pd复合物,与氰甲基Cu2+发生金属转移,进而释放金属Pd生成C(sp3)—H键乙腈化产物(31)。该反应借助分子内的导向基团调控与金属Pd复合激活潜在C—H键,进而成功构建了一种直接发生氰甲基化的方法。
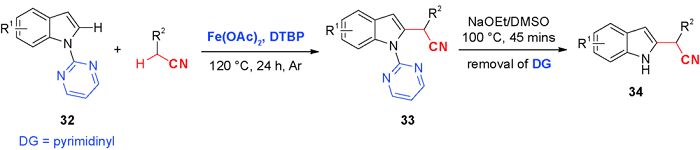
Guo等[31]报道了一种吲哚或吡咯分子(32)C-2位发生氰甲基化反应,该反应利用含二价醋酸铁催化,嘧啶基作为导向基团,成功构建了sp3-sp2氧化偶联反应,制备一系列乙腈衍生物(33)。研究表明导向基团的嘧啶分子中的N原子与活化的含铁原子配位,进而氰甲基自由基通过加成进攻,释放二价铁生成氰甲基产物。该反应需要自由基引发剂以及高温120 ℃来完成。且导向基团的嘧啶基可以在NaOEt/DMSO高温下很容易除去得到一系列2-乙腈取代吲哚衍生物(34)。
综上,直接利用乙腈或取代乙腈进行氰甲基化主要是自由基策略制备高活性的氰甲基自由基,进而对底物分子中的双键、芳烃、甚至是未激活的C(sp3)—H键,借助金属配位催化或导向基团诱导活化等方法构建C—C键形成反应,进而发生一系列的转化制备氰甲基产物。
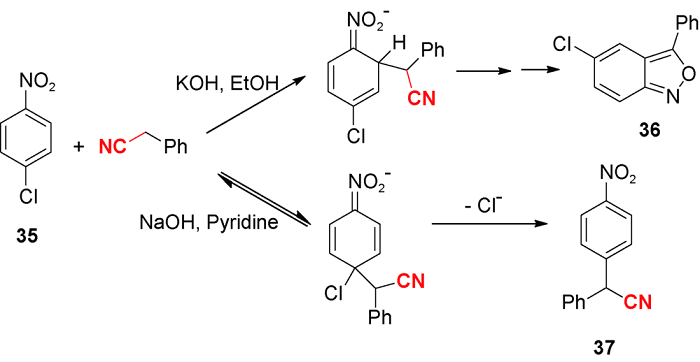
Pizzini等[32]最早报道硝基烯烃与苯乙腈碳负离子的反应,当在KOH的乙醇水溶液中,反应得到5-氯-3-苯基苯并口恶唑(36),而在非质子溶剂吡啶存在下,亲核取代SNAr反应发生,制备得到芳基乙腈取代产物(37)。随后Mᶏkosza等[33]在2004年对缺电子芳烃或杂环发生亲核取代ArH-CH反应进行了详细的评述。其中,关于亲核取代芳基C—H进行氰甲基化的机理进行了系统的阐述,认为缺电子芳烃特别是含硝基的芳烃,可以和亲核试剂发生加成反应生成δH-adducts,该加成体可以发生快速转化反应,生成亲核取代芳基C—H产物,他们定义这个过程为oxidative nucleophilic substitution of hydrogen(ONSH)[34],并且这个反应可以应用到各种有机合成反应中。明显地,这也是一种芳环氰甲基化方法,然而随后的文献中却并未得到深入的关注。
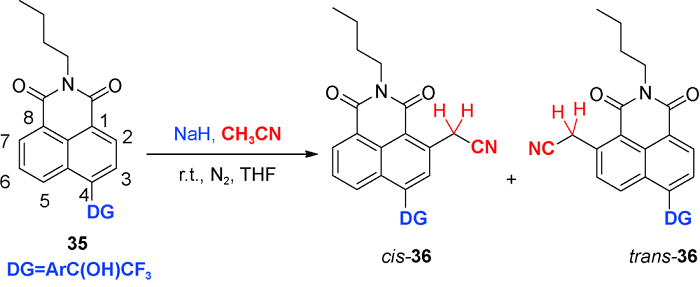
上述Mᶏkosza等工作引起本课题组强烈的兴趣,在创制新颖阴离子受体以及对荧光团的结构改造过程中,我们对缺电子的荧光团构建含CHCN作用位点以及发展潜在的氰甲基化方法做了一些探索性工作[35,36]。特别是,在构建1,8-萘酰亚胺荧光团用来设计化学传感器及生物探针材料方面[37,38],修饰1,8-萘酰亚胺的常用方法是在C3、C4或C5位引入一个官能团[39,40],进而取得强烈的荧光信号用于检测环境及细胞中的待测分子,然而1,8-萘酰亚胺中的C2及C7位置很少被利用。且在C2或C7位引入CH2CN可能与1,8-萘酰亚胺的羰基形成分子内C=O…H-CCN氢键。因此,我们成功地在C4位引入三氟甲基吡啶基甲醇基作为导向基团或拉电子基团来辅助芳环C—H键的活化(35),通过乙腈碳负离子对芳环C—H的亲核取代,进而实现C2或C7位的氰甲基化,从而得到具有顺反异构特性的氰根离子探针cis-36和trans-36[41]。
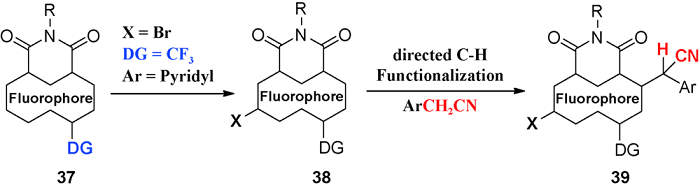
进一步,将-CF3作为导向基团并用于强化芳环的缺电子性,引入1,8-萘酰亚胺芳环的C4-位(37),进而对荧光团骨架上的C2或C7位置的C—H官能化进行激活,并且通过在C6位置引入卤素溴(38)来抑制反应产生官能团位置异构,专一且高效地在C2位置发生氰甲基化。相对于之前的工作,该合成策略具有更好的区域选择性,且反应条件温和,只需要NaH催化,室温即可发生萘酰亚胺C2位的直接芳乙基化,并且产物(39)分子[42]中吡啶环和羰基能够有效稳定探针39与阴离子作用后产生的碳负离子,进而使探针39对阴离子发挥较好的识别效果。
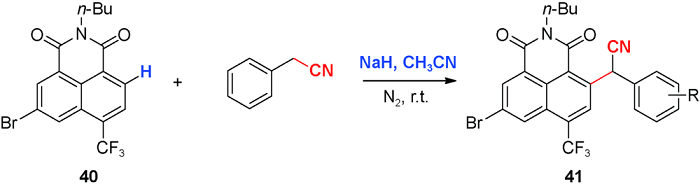
进一步对该反应的方法学进行详细的研究[43],以萘酰亚胺(40)与苯乙腈为模型反应,详尽地探讨了反应浓度、碱当量、反应时间等因素,最终在NaH、乙腈条件下,室温6 h即可方便地发生C—H芳乙腈化,该反应条件温和,特别是不需要过渡金属催化的条件下,就可以得到一系列α-二芳基乙腈衍生物(41),拓宽了萘酐染料或发色团的结构多样性。
6 结论及展望
本文主要归纳总结近年来有关CH氰甲基/烷基化反应以及本课题组在1,8-萘酐荧光团骨架上发展的C—H氰甲基化反应并成功应用到阴离子检测。总结下来研究最多的策略是通过自由基引发剂或光照生成稳定的氰甲基自由基,进而对底物分子中的双键进行C—C键生成,进而得到目标产物。利用导向基团策略进行氰甲基化的研究不多,因此,继续探索导向基团策略构建有用的C—H氰甲基化反应,拓宽底物的适用性,发展高效且绿色的策略用于发现更多优美的功能分子应用到药物或探针领域中,将是合成化学领域有趣且重要的研究方向。