




1 引言
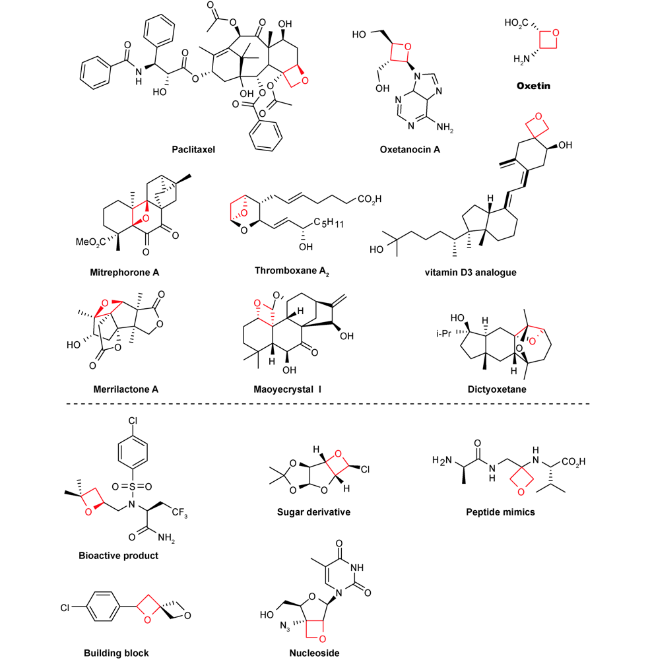
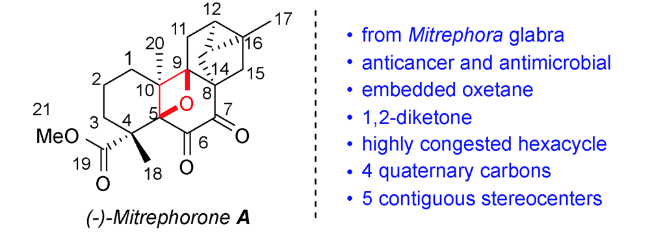
在药物化学领域里,氧杂环丁烷结构常用作偕二甲基和羰基的替代基团以改善母体分子的物理化学性质[11⇓~13]。在有机合成领域里,氧杂环丁烷因其自身具有较大环张力的特点,可发生亲核开环、重排、扩环等官能团转换反应构建结构多样性的有机化合物[14⇓⇓⇓⇓⇓~20]。在天然产物中,氧杂环丁烷骨架更是呈现出独特的生物活性。以紫杉醇为代表的含氧杂环丁烷结构的天然产物在人类抗击癌症和推动有机化学发展的历史上发挥了重要作用,紫杉醇复杂的环系及优秀的生物活性引起合成化学家和药物化学家的极大兴趣。1971年,科学家从西太平洋紫衫(红豆衫属)的茎皮分离出Taxol,发现其可用于癌症治疗。通过计算等研究发现,在紫衫醇结构中,氧杂环丁烷结构表现出构像锁定、结构稳定和氢键受体的作用[21,22]。后来,人们相继从土壤细菌、链霉菌、菠菜叶子中分离得到多种含氧杂环丁烷片段的具有抗HIV、抗癌、抑制谷氨酰胺合成酶等作用的天然产物Paclitaxel(紫杉醇,也称泰素)、Oxetin(氧丁霉素)、Oxetanocin A、Mitrephorone A、Thromboxane A2(血栓素A2)、vitamin D3(维生素D3)类似物、Merrilactone A、Maoyecrystal I、Dictyoxetane等(图1)[23⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓~40]。
氧杂环丁烷作为含氧的饱和四元杂环,具有106 kJ/mol的环张力,比氧杂环丙烷的环张力能量112 kJ/mol略小。通过单晶衍射分析,发现其具有近似平面结构,由于氧原子的存在,明显减少了结构中的邻位交叉作用(guache interaction),这种结构弱化了其环张力[41,42]。另外,氧上孤对电子的存在,可使氧杂环丁烷化合物作为路易斯碱和氢键受体。正因为以上这些独特结构,氧杂环丁烷常作为偕二甲基和羰基的生物电子等排体,可改善药物骨架的物理化学和药代动力学性质[43,44]。例如,2006年,Carreira等[11]就报道了3,3-二取代氧杂环丁烷在药物化学领域具有替代偕二甲基的前景。发现氧杂环丁烷片段的引入,规避了偕二甲基过度亲脂性的缺点,提高了药物可溶性、药代动力学性质和代谢稳定性;另外对于药物中含有的羰基化合物(醛酮酯),由于其在人体中有易受酶攻击、去质子化和手性中心消旋化的缺陷。若引入不同链长取代基的氧杂环丁烷结构,就会改善这一结果,并且还能增加药物与靶细胞的亲合力。除此之外,氧杂环丁烷片段还存在对б电子的吸电子作用,可改善母体化合物的酸碱性[45⇓⇓⇓⇓~50]。
探索氧杂环丁烷的合成方法具有重要意义。2016年,Morgan等在Chemical Review上发表的综述介绍了2010~2015年这五年内关于氧杂环丁烷的合成、反应活性及药物化学领域的进展,同时也提供了之前的总结性文章[6]。本文将按成键方式分类,综述2016~2020年这五年关于氧杂环丁烷合成方法的最新成果,包括C—C键形成的环化反应、C—O键形成的环化反应、[2+2]环加成反应、环氧乙烷扩环反应、C—H键氧化环化反应来系统地总结该领域的进展,包括每类方法在天然产物及药物合成中的应用,为有机合成同行尤其是对研究氧杂环丁烷合成及应用的学者提供一些有价值的信息。
2 C—C键形成的分子内环化反应
通过由C—C键形成来构建氧杂四元环结构的方法近年来报道得很少,常规方法主要还是以分子内取代反应为主,未见有新颖方法的报道。本文将择选个别2016年前的报道再结合最近五年的发展,叙述该类方法的最新成果。
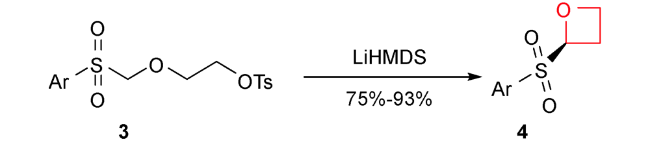
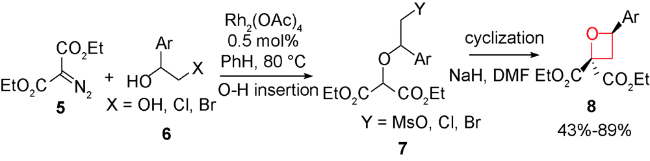
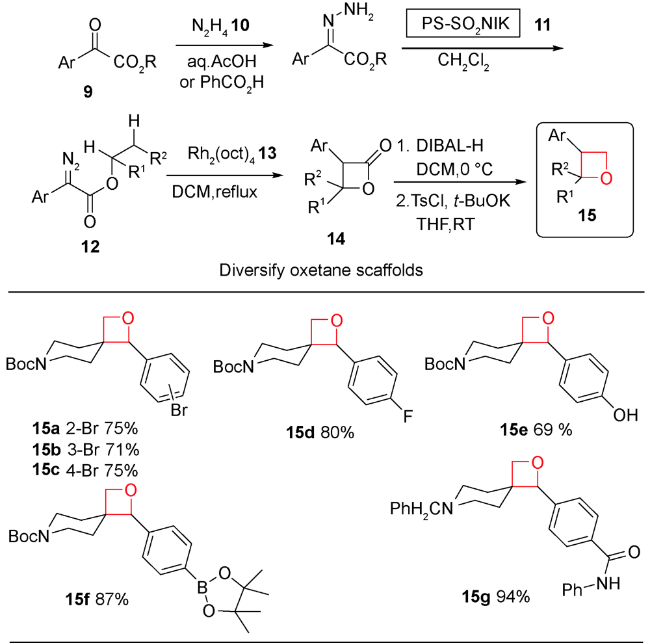
2017年,Moody等报道了重氮酸酯在金属铑催化下发生铑卡宾C—H插入形成β-丙内酯,然后还原得到螺环氧杂环丁烷化合物[53]。从简单易得的酮酯9出发,先与肼10反应形成腙,再经过含有氧化剂PS-TsNIK 11的填充柱发生重氮化得到卡宾前体12。然后,不需分离,二价铑13存在下二氯甲烷中回流,发生选择性分子内C—H键插入反应,以32%~88%的产率得到结构多样性的螺环β-丙内酯类化合物14。接着发生DIBAL-H低温还原得到44%~91%产率的多样性3-螺环氧杂环丁烷产物15。整个反应过程操作简便,重氮化合物无需分离,底物普适性较好且区域选择性专一。通过对螺环氧杂环丁烷化合物的官能团转化,得到一系列结构新颖的含氧杂环丁烷骨架的化合物15a~15g(图式3)。
通过分子内碳碳键形成的环化反应构建氧杂环丁烷的合成方法,主要围绕过渡金属催化的O—H插入,然后通过亲核取代环化。或者先制备重氮乙酸酯环化前体,然后借助过渡金属催化的C—H键插入及还原实现多取代四元环骨架的构建。但高效且不对称合成多取代氧杂环丁烷化合物一直是该类方法的巨大挑战,如何实现高效高对映选择性、底物普适性更广的新颖C—C键形成方法来合成氧杂环丁烷仍是该方法值得探索的目标。
3 C—O键形成的分子内环化反应
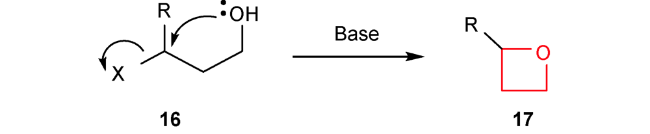
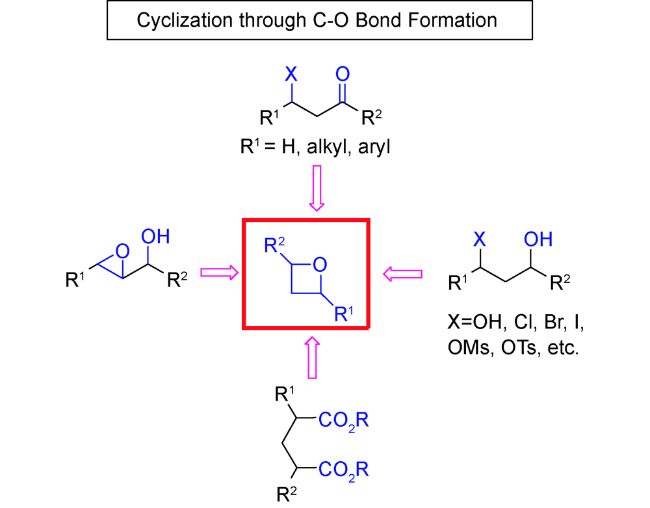
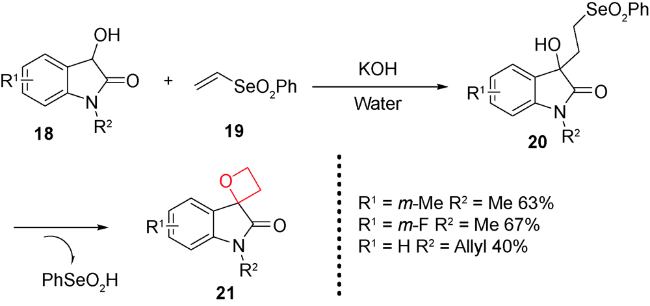
随着过渡金属不对称催化的发展,该方法显示出原子经济性较好、高效等独特的优势。因此,有机合成化学家们也尝试通过金属催化C—O键形成的分子内环化反应来合成结构新颖的氧杂环丁烷类化合物。以烯丙或炔丙醇为底物,利用过渡金属活化碳碳双键和三键,形成类似于卡宾中间体的电子受体,然后再发生环化反应得到四元环骨架。
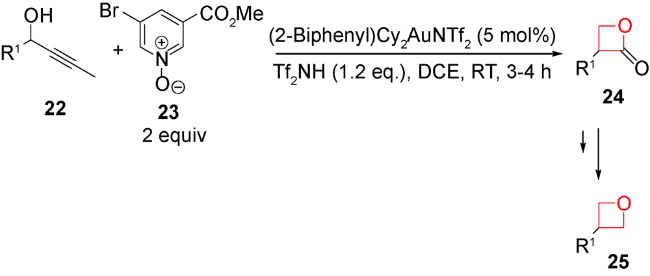
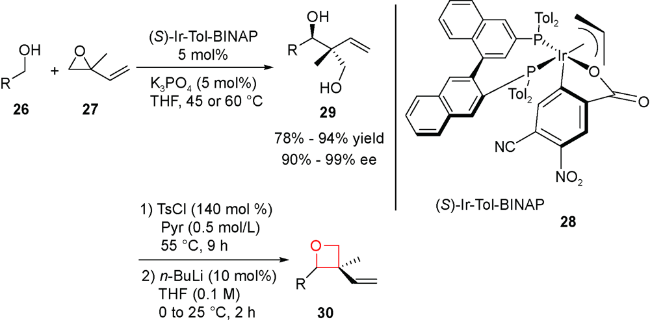
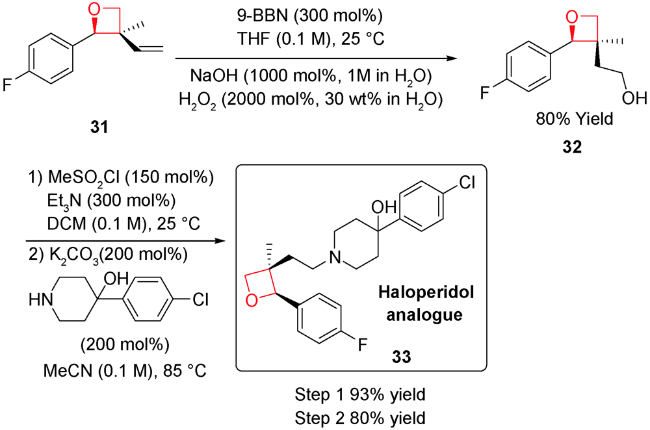
另外,实现该类方法的不对称反应也是目前的发展方向。但主要挑战在于如何高立体化学专一得到γ-二醇或其类似物,另外,在环化过程中如何控制立体化学,避免消旋化。2016年,Krische等设计合成了一类刚性结构的铱催化剂28,完成了首例含季碳中心的螺环氧杂环丁烷的不对称合成[63]。
4 氧杂环丙烷扩环反应
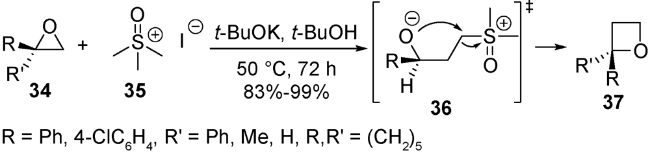
扩环方法学是合成杂环化合物的重要方法之一,氮及硫杂三元环扩环成四元环起步较晚,已受很多人关注[65⇓⇓⇓⇓⇓⇓⇓⇓~74]。氧杂三元环扩环成四元环发现较早,也已相对发展得更加成熟。1979年,Welch等报道了亚砜肟阴离子作为亚甲基转移试剂与酮在二甲基亚砜中40 ℃条件下反应,经过氧杂环丙烷中间体发生扩环得到氧杂环丁烷。但该方法存在原料制备繁琐及效率不高的问题[75]。1983年,Ohta等通过调整亚甲基转移试剂,发展了用常见的二甲基亚砜甲基叶立德35作为试剂,在一锅条件下与由醛酮34经过Corey-Chaykovsky环氧化反应得到的氧杂环丙烷36原位发生扩环反应,直接得到2-取代和2,2-二取代氧杂环丁烷37(图式10)。该方法具有简单高效、条件温和、底物耐受较广的特点[76]。
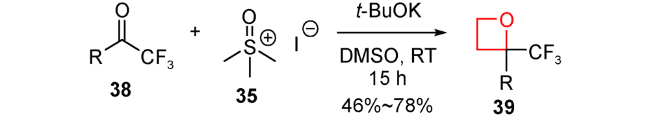
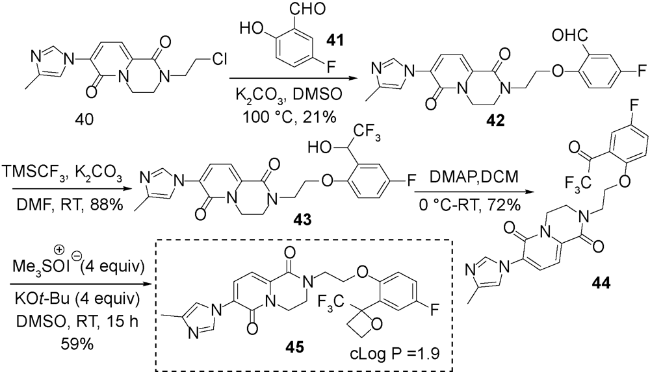
近年来,随着氟化学的发展,人们认识到,当药物分子引入氟原子,会改变药物的亲脂性、氢键作用、代谢稳定性、生物利用度等,可提高药物的活性[81]。2017年,am Ende等发现用于治疗阿尔茨海默病的γ-分泌酶调节剂引入2-三氟甲基氧杂环丁烷片段可改善药物的亲脂性,具有潜在的应用前景[82]。当时对于含有强吸电子取代基如三氟甲基的氧杂环丁烷底物的合成方法还未见报道,就选择三氟甲基酮类化合物38作为底物,二甲基亚砜甲基叶立德作为亲核性的亚甲基转移试剂35,在温和条件得到46%~78%产率且底物耐受性较好的2-三氟甲基氧杂环丁烷类化合物39(图式11)。另外,还利用该方法合成了一种γ-分泌酶调节剂类似物45(图式12),该分子经过生物活性测试表现出不错的物理化学性质及药物活性。
由氧杂环丙烷扩环反应合成氧杂环丁烷的方法已发展较为成熟,其是合成2,2-二取代氧杂环丁烷的重要方法之一,不管是调节亚甲基转移试剂硫叶立德,还是在价廉易得的酮底物进行底物结构修饰方面,都表现出不错的应用价值。但如何开发出更多的不对称催化合成光学活性的氧杂环丁烷的方法并应用到天然产物或药物分子的合成中,也是其接下来探索的重要方向之一。
5 [2+2]环加成反应
5.1 Paternò-Büchi [2+2]光反应
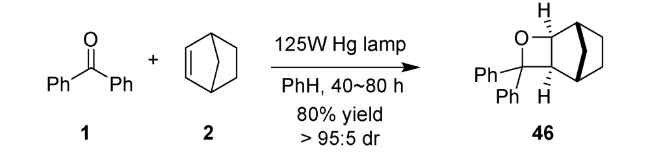
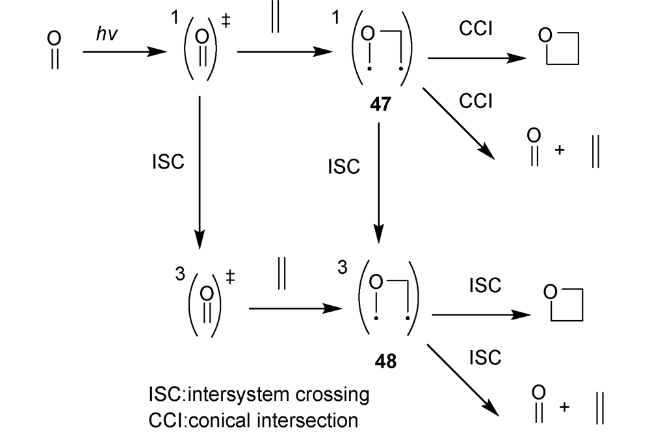
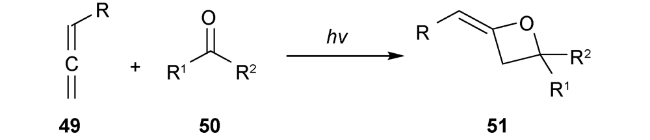
自1909年 Paternò 和Chieffi发现了第一例光介导羰基化合物1与烯烃化合物2环加成得到氧杂环丁烷化合物46以来(图式13),对该反应的研究就一直未间断[2]。为确定产物结构为氧杂环丁烷衍生物,科学家就探索近半个世纪。1954年,Büchi等重复实验后确证为氧杂四元环结构[3]。1964年,Yang等首次发现了其化学选择性关环的普遍规律[4],此后Paternò-Büchi 反应便闻名于世。该反应的机理研究发现主要涉及双自由基中间体47[83]。酮羰基经过光照激发及系间窜越生成三线态,接着与C=C双键作用,得到三线态自由基中间体48,然后快速经过系间窜越得到单线态双自由基中间体,进而发生环化构建四元环或断裂C—O键回到原料(图式14)。Paternò-Büchi反应具有明显的底物限制,越有利于双自由基中间体稳定,烯烃活性越高,则有利于环化得到氧杂四元环化合物。该反应适用于芳香酮类或醛类化合物与富电子烯烃来构建氧杂环丁烷结构。对于烷基酮类底物则大多需要超高压辐照和石英反应器,不具备实用性和操作友好性。
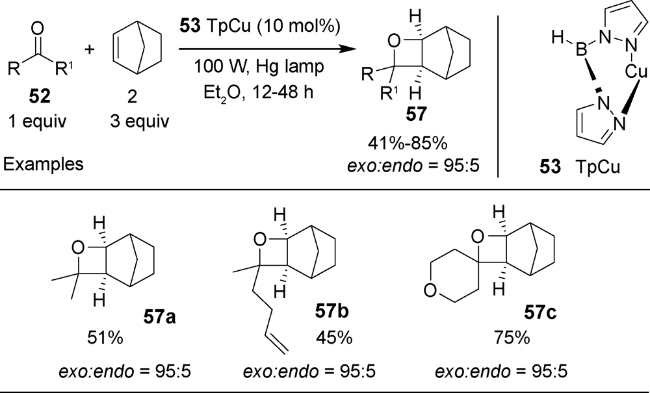
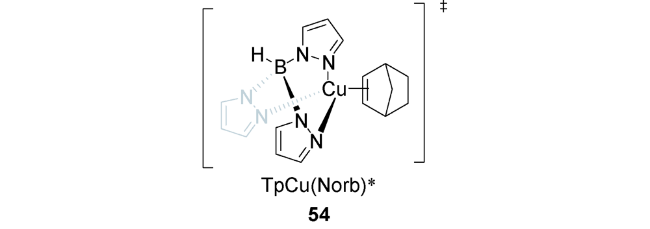
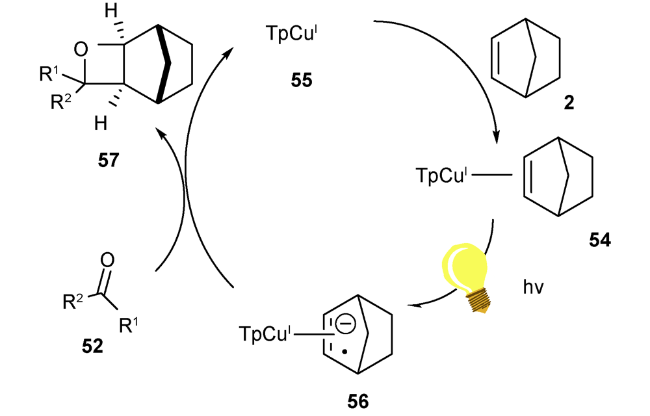
2019年,Schmidt等利用过渡金属催化的方法,由中心金属与配体在光照辐射条件下发生电荷转移来活化烯烃,得到活性金属物种,后者再被烷基醛酮羰基原位捕获,得到[2+2]的氧杂环丁烷产物(图式16)[85]。最佳反应条件为1∶3配料比的酮52与降冰片烯2,在10 mol%的三吡唑硼铜(Ⅰ)络合物(TpCu)53的催化下,在乙醚溶剂中100 W汞灯光照条件下,经过12~48 h可得41%~85%产率的高选择性(exo: endo > 95∶5)的目标化合物。值得一提的是,体系生成的TpCu-(Norb) 54是重要的光活性物种,对反应顺利发生起着至关重要的作用。该反应的机理为TpCuI化合物55与烯烃2配位形成TpCu(Norb)54( 图2),后者在280~300 nm光照条件下,发生金属与配体的电荷转移,得到Cu(Ⅱ)金属中心和烯烃的自由基阴离子物种56,然后该极化中间体随即被羰基捕获,得到目标化合物57(图式17)。
5.2 形式[2+2]环加成反应
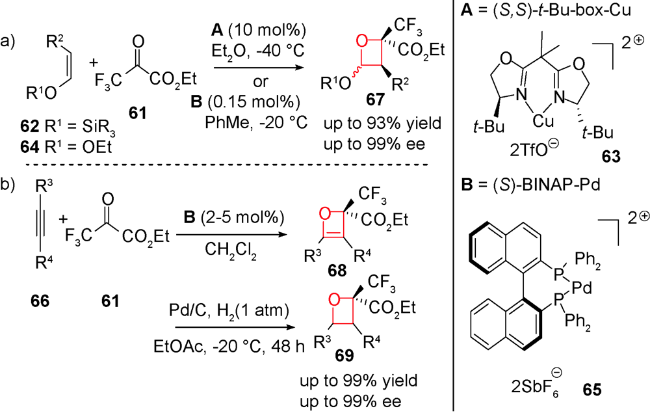
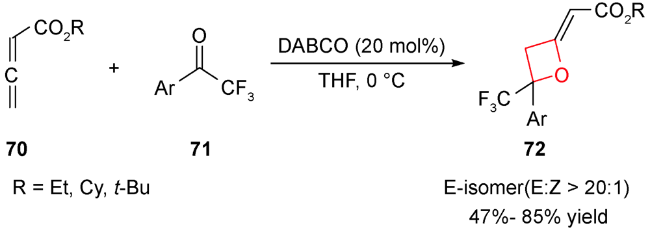
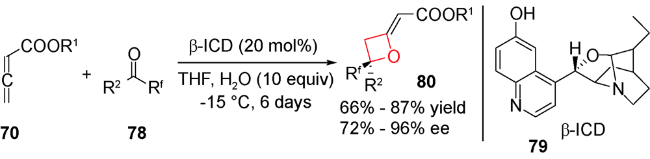
除了路易斯酸催化,最近还报道了路易斯碱催化发生的形式[2+2]环加成反应。2011年,Ye等用DABCO催化了该类环加成反应。无取代的累积二烯酸酯70与三氟甲基酮71在20 mol%的DABCO催化下,生成高非对映选择性和中等产率的2-烯基氧杂环丁烷化合物72(图式20)[89],机理如图式21所示:手性叔胺73与累积二烯键选择性加成得到两性离子中间体74和75,后者再与三氟甲基酮71发生加成,得到中间体季铵盐76,再发生分子内环化得到中间体77,最后发生季铵盐消除释放叔胺催化剂,并得到目标化合物。在此基础上,2012年Shi等利用手性叔胺实现了累积二烯酸酯70与三氟甲基酮78的不对称形式[2+2]环加成反应(图式22)[90]。通过对手性胺催化剂的筛选,发现在20 mol% β-ICD 79,10当量水和低温条件下,可顺利实现该反应,并取得了不错的产率及ee值,该体系中过渡态存在的氢键对对映选择性控制产生至关重要的作用。
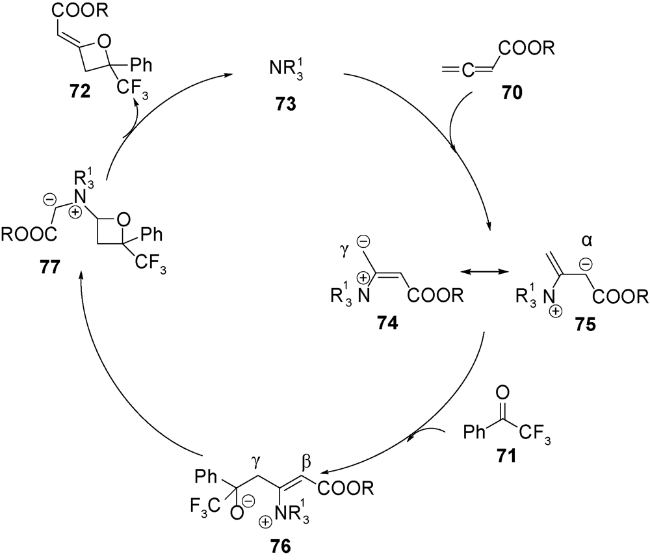
2013年和2015年,Chi等和Smith等分别报道了利用氮杂环卡宾可以实现不对称催化醛和三氟甲基酮[2+2]环加成生成含有氧杂环丁烷结构的不饱和酯类化合物,后者再经过还原可得到结构多样化且具有光学活性的氧杂环丁烷化合物[91,92]。受此启发,2018年,Scheidt等首次报道了氮杂环卡宾(NHC)81催化的γ-取代的累积二烯酸酯82与三氟甲基酮83发生形式[2+2]环化反应,可一步得到具有优秀产率和良好非对映选择性的2-烯基取代的氧杂环丁烷84和85(图式23)[93]。进一步将86烯烃碳碳双键进行官能团转换得到多样性的氧杂环丁烷化合物87和88(图式24)。系统的机理研究发现,氮杂环卡宾催化剂89在累积二烯烃的β碳位置进攻,发生亲核加成得到两性离子中间体90,后者再与酮发生γ位的亲核加成得到中间体91,接着氧负离子进攻化合物91的β碳得到烯醇中间体92,最后NHC经过还原消除再生,并释放出目标化合物,从而完成整个催化循环(图式25)。
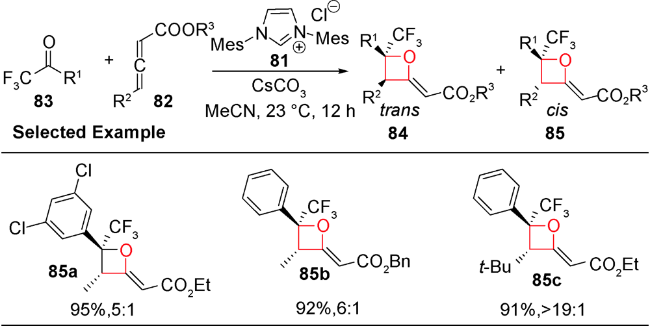
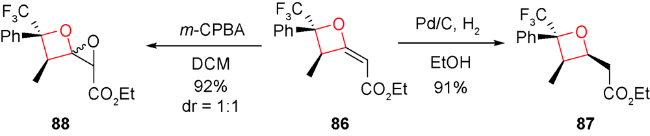
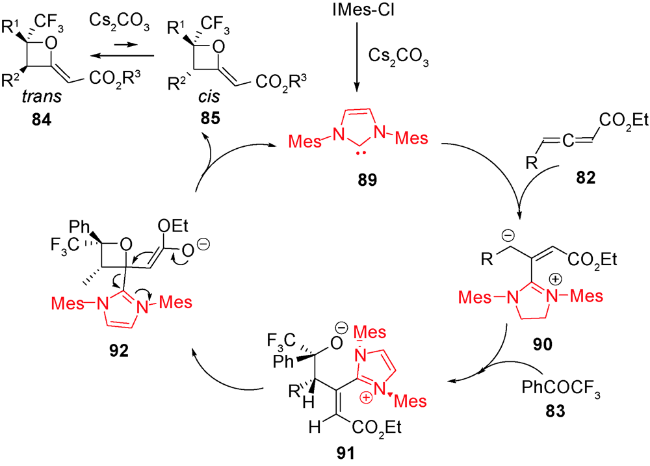
酸碱催化的形式[2+2]环加成反应具有操作简单、高效高选择性、底物官能团耐受性较好的特点,具有很大的发展潜力。
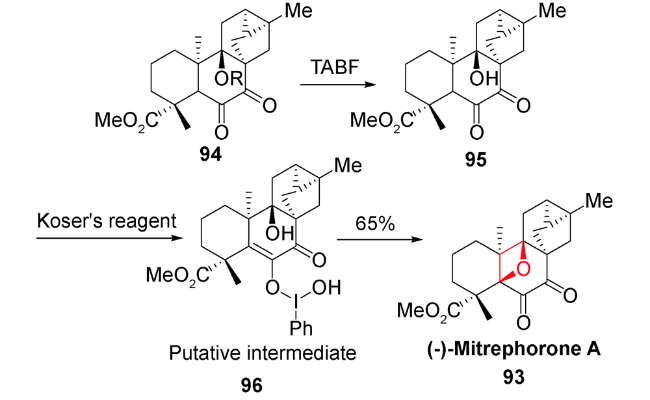
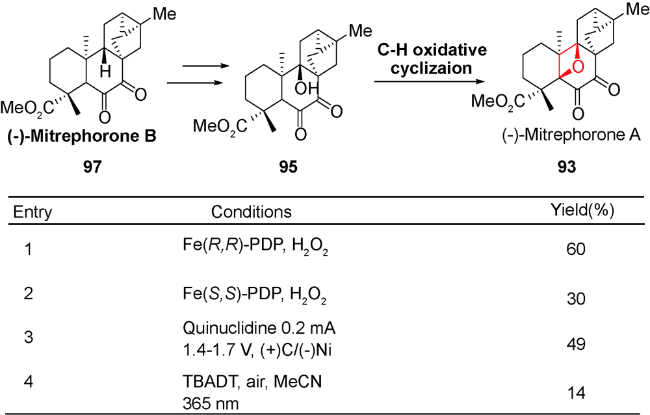
6 C—H键氧化环化反应
正如前文叙述,含氧杂环丁烷的结构广泛存在于天然产物中,例如紫杉醇、oxetanocin A、oxetinoxetin、merrilactone A等,其中氧杂环丁烷的引入是合成策略的核心。而依靠常规的扩环、[2+2]环加成反应和亲核取代反应等形成氧杂环丁烷的方法存在很大的底物局限性。如何在天然产物中实现该核心片段的构建一直是具有挑战性的工作。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
通过对氧化剂的筛选和结构相关底物的研究,发现该氧化步骤的产率都不高,说明目前的C—H键氧化环化方法仍存在局限性和发展空间,生源合成途径也需要进一步的研究。
7 结论
氧杂环丁烷是一种饱和的氧杂小环化合物,其作为结构单元在天然产物、药物化学、材料化学及有机合成中发挥着重要的作用。本文较系统综述了近五年氧杂环丁烷的合成方法学,对研究氧杂四元环结构骨架的构建具有重要的指导意义。纵观近一百多年来关于氧杂环丁烷类化合物的合成方法发展史,在目前已有的通过C—C键形成的环化反应、C—O键形成的环化反应、[2+2]环加成反应、扩环反应及C—H键氧化环化反应等方法中,通过C—C键和C—O键形成的环化反应发展得最早,也较成熟。而[2+2]环加成反应、扩环反应及C—H键氧化环化反应等方法具有较大的研究空间,因为该类方法都普遍存在底物受限、步骤繁多、不对称方法较少等缺点。近年来,随着光化学、电化学及仿生合成等领域的蓬勃发展,如何结合新合成方法的特点及优势来拓宽氧杂环丁烷的不对称合成方法学,克服以上局限将是接下来有机合成工作者致力研究的目标。