




1 引言
2 用于氧化降解EOCs的Fe-MOFs及其复合物的制备
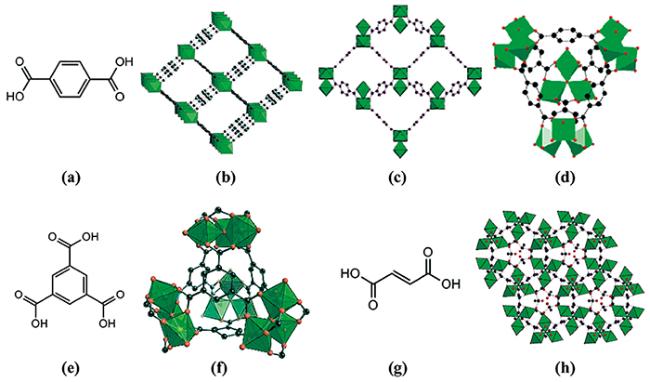
图1 各羧酸配体图和Fe-MOFs结构图:(a) 对苯二甲酸;(b) MIL-53(Fe)[42];(c) MIL-88B(Fe)[42];(d) MIL-101(Fe)[42];(e) 均苯三甲酸;(f) MIL-100(Fe)[44];(g) 富马酸;(h) MIL-88A(Fe)[43]Fig.1 Diagrams of carboxylic acid ligands and Fe-MOFs structure:(a) 1,4-dicarboxybenzene;(b) MIL-53(Fe)[42];(c) MIL-88B(Fe)[42];(d) MIL-101(Fe)[42];(e) 1,3,5-benzenetricarboxylic acid;(f) MIL-100(Fe)[44];(g) fumaric acid;(h) MIL-88A(Fe)[43] |
2.1 MIL-100(Fe)及其复合物
表1 铁基MOFs及其复合材料对EOCs的降解性能Table 1 The degradation performance of iron-based MOFs and their composites for EOCs |
| Catalysts/Dosage (g·L-1) | Target Pollutants/Volume (mL)/concentration (mg·L-1)/pH | Light Source | Reaction Time (min) | Degradation Efficiency (%) | ref |
|---|---|---|---|---|---|
| Photocatalytic oxidation | |||||
| WO3/MIL-53(Fe)/0.2 | 2,4-dichlorophenoxyacetic acid/100/45/2.5 | sun light | 240 | ~100 | 45 |
| CdS/MIL-53(Fe)/0.75 | ketorolac tromethamine/100/10/6 | 85 W Oreva CFL bulb(λ≥ 420 nm) | 330 | 80 | 46 |
| MIL-88A/g-C3N4/1.0 | tetracycline/100/10/NA | 1000 W iodine tungsten lamp(λ≥ 420 m) | 120 | 22 | 47 |
| Ag/AgCl@MIL-88A(Fe)/0.4 | ibuprofen/50/10/NA | 500 W Xe lamp(λ≥ 420 nm) | 210 | 100 | 48 |
| BiOI/MIL-88B(Fe)/0.3 | ciprofloxacin/100/10/NA | 150 W Xe lamp(AM 1.5G) | 270 | 80 | 49 |
| MIL-100(Fe)/PANI/0.25 | tetracycline/200/10/NA | 300 W Xe lamp | 120 | 84 | 36 |
| Fenton-like reaction | |||||
| 1T-MoS2@MIL-53(Fe)+20 mmol/L·H2O2/0.4 | ibuprofen/50/10/7.0 | 500 W Xe lamp(λ≥ 420 nm) | 120 | 100 | 50 |
| g-C3N4/PDI@NH2-MIL-53(Fe)+10 mmol/L·H2O2/0.4 | tetracycline/50/50/6.0 | 5 W LED white lamp(380~800 nm) | 40 | 90 | 51 |
| g-C3N4/PDI@NH2-MIL-53(Fe)+10 mmol/L·H2O2/0.4 | carbamazepine/50/50/6.0 | 5 W LED white lamp(380~800 nm) | 150 | 78 | 51 |
| g-C3N4/PDI@NH2-MIL-53(Fe)+10 mmol/L·H2O2/0.4 | bisphenol A/50/50/6.0 | 5 W LED white lamp(380~800 nm) | 10 | 100 | 51 |
| g-C3N4/PDI@NH2-MIL-53(Fe)+10 mmol/L·H2O2/0.2 | bisphenol A/50/2/6.0 | 5 W LED white lamp(380~800 nm) | 10 | 100 | 51 |
| MIL-88A(Fe)+100 μL H 2O2/0.2 | bisphenol A/50/10/NA | 350 mW LED visible light | 60 | ~100 | 52 |
| PANI/MIL-88A(Fe)+20 μL H 2O2/0.2 | bisphenol A/50/10/5.1 | 5 W LED visible light | 30 | 100 | 53 |
| CUS-MIL-100(Fe)+6 mmol/L H2O2/0.5 | sulfamethazine/80/20/3.0 | in dark | 60 | 100 | 17 |
| Pd@MIL-100(Fe)+40 μL H 2O2/0.125 | theophylline/40/20/4.0 | 300 W Xe lamp(λ≥ 420 nm) | 150 | 99.5 | 54 |
| Pd@MIL-100(Fe)+40 μL H 2O2/0.125 | ibuprofen /40/20/4.0 | 300 W Xe lamp(λ≥ 420 nm) | 150 | 100 | 54 |
| Pd@MIL-100(Fe)+40 μL H 2O2/0.125 | bisphenol A/40/20/4.0 | 300 W Xe lamp(λ≥ 420 nm) | 150 | 68 | 54 |
| Pd-PTA-MIL-100(Fe)+40 μL H 2O2/0.125 | theophylline/40/20/4.0 | 300 W Xe lamp(λ≥ 420 nm) | 150 | 99.5 | 55 |
| Pd-PTA-MIL-100(Fe)+40 μL H 2O2/0.125 | ibuprofen/40/20/4.0 | 300 W Xe lamp(λ≥ 420 nm) | 180 | 99.5 | 55 |
续表1 铁基MOFs及其复合材料对EOCs的降解性能Table 1 (continued) The degradation performance of iron-based MOFs and their composites for EOCs |
| Catalysts/Dosage (g·L-1) | Target Pollutants/Volume (mL)/concentration (mg·L-1)/pH | Light Source | Reaction Time (min) | Degradation Efficiency (%) | ref |
|---|---|---|---|---|---|
| WO3/MIL-100(Fe)+40 μL H 2O2/0.25 | bisphenol A/80/10/3.0 | 25 W LED visible light | 20 | 100 | 56 |
| MIL-100(Fe)/g-C3N4+50 μL H 2O2/0.5 | diclofenac sodium/200/0.1 mmol/L/NA | 300 W Xe lamp | 50 | 100 | 21 |
| MIL-100(Fe)/Fe-SPC+40 mmol/L·H2O2/1.0 | thiamethoxam/50/60/7.5 | 600 W ultrasonic probe | 100 | 100 | 57 |
| Cu2O/MIL-100(Fe/Cu)+49 mmol/L H2O2/0.5 | thiacloprid/50/80/7.47 | 500 W Xe lamp | 25 | 90 | 58 |
| MIL-100(Fe)/TiO2+20 μL H 2O2/0.05 | tetracycline /100/100/NA | 450 W Xe arc lamp | 60 | 85.8 | 59 |
| MIL-100(Fe)@Fe3O4/CA+H2O2/0.2 | tetracycline /50/10/5.0 | 150 W Xe lamp(λ≥ 400 nm) | 210 | 85 | 60 |
| M.MIL-100(Fe)@ZnO +10 mmol/L·H2O2/0.2 | bisphenol A/50/5/2.0 | LSH-500 W Xe arc lamp | 60 | ~100 | 61 |
| M.MIL-100(Fe)@ZnO +10 mmol/L·H2O2/0.2 | atrazine/50/5/2.0 | LSH-500 W Xe arc lamp | 120 | >80 | 61 |
| Oxidation of activated persulfate | |||||
| AgIO3/MIL-53(Fe)+50 mg·L-1 PS/0.5 | methyl malathion/100/20/5.0 | sun light | 120 | 93 | 37 |
| AgIO3/MIL-53(Fe)+50 mg·L-1 PS/0.5 | chlorpyrifos/100/20/5.0 | sun light | 120 | 97 | 37 |
| AgIO3/MIL-53(Fe)+50 mg·L-1 PS/0.5 | methyl malathion(binary mixture)/100/20/5.0 | sun light | 180 | 100 | 37 |
| AgIO3/MIL-53(Fe)+50 mg·L-1 PS/0.5 | chlorpyrifos(binary mixture)/100/20/5.0 | sun light | 180 | 50 | 37 |
| MIL-88A@MIP+10.8 mmol/L PS/0.5 | dibutyl phthalate/100/3.5/ not adjusted | in dark | 480 | 77.4 | 62 |
| MIL-88A@MIP+10.8 mmol/L PS/0.5 | dibutyl phthalate/100/4.0/ not adjusted | in dark | 480 | >80.4 | 62 |
| MIL-88A@MIP+10.8 mmol/L PS/0.5 | dibutyl phthalate/100/5.0/ not adjusted | in dark | 480 | 80.4 | 62 |
| MIL-88B(Fe)+2 mmol/L PS/0.6 | bisphenol A/100/10/6.5~7.2 | 300 W Xe lamp(λ≥ 420 nm) | 25 | 100 | 63 |
| Bi12O17Cl2/MIL-100(Fe)+0.2 mmol/L PS/0.25 | bisphenol A/200/10/5.2 | 300 W Xe lamp | 40 | 100 | 64 |
| g-C3N4/MIL-101(Fe)+1 mmol/L PS/0.5 | bisphenol A/NA/10/NA | 300 W Xe lamp(λ≥ 400 nm) | 60 | 98 | 34 |
| AQS-NH-MIL-101(Fe)+10 mmol/L PS/0.2 | bisphenol A/25/60/5.76 | in dark | 120 | 97.7 | 35 |

图2 (a) Pd/MIL-100(Fe)[54]、(b) Pd-PTA-MIL-100(Fe)[55]、(c) CUS-MIL-100(Fe)[17]、(d) MIL-100(Fe)/Fe-SPC[57]、(e) Cu2O/MIL(Fe/Cu)[58]、(f) Cu2O/MIL(Fe)[58]、(g) MIL-100(Fe)/Ti2[59]、(h) M.MIL-100(Fe)@ZnO[61]和(i) MIL-100(Fe)@Fe3O4/CA[60]的形貌图Fig.2 The morphologies of (a) Pd/MIL-100(Fe)[54],(b) Pd-PTA-MIL-100(Fe)[55],(c) CUS-MIL-100(Fe)[17],(d) MIL-100(Fe)/Fe-SPC[57],(e) Cu2O/MIL(Fe/Cu)[58];(f) Cu2O/MIL(Fe)[58];(g) MIL-100(Fe)/Ti2[59];(h) M.MIL-100(Fe)@ZnO[61] and (i) MIL-100(Fe)@Fe3O4/CA[60] |

2.2 MIL-101(Fe)及其复合物

2.3 MIL-53(Fe)及其复合物

2.4 MIL-88(Fe)及其复合物

图6 (a) MIL-88A-1[52]、(b) MIL-88A-2[52]、(c) PANI/MIL-88A(Fe)[53]、(d) MIL-88A/g-C3N4[47]、(e) MIL-88A@MIP[62]、(f) Ag/AgCl@MIL-88A(Fe)[48]、(g) MIL-88B(Fe)[63]、(h) MIL-88B(Fe)[49]和(i) BiOI/MIL-88B(Fe)[49]的形貌图Fig.6 The morphologies of(a) MIL-88A-1[52],(b) MIL-88A-2[52],(c) PANI/MIL-88A(Fe)[53],(d) MIL-88A/g-C3N4[47],(e) MIL-88A@MIP[62],(f) Ag/AgCl@MIL-88A(Fe)[48],(g) MIL-88B(Fe)[63],(h) MIL-88B(Fe)[49] and (i) BiOI/MIL-88B(Fe)[49] |
3 Fe-MOFs及其复合物高级氧化降解EOCs的应用
3.1 Fe-MOFs及其复合物高级氧化降解水中药物
3.2 Fe-MOFs及其复合物高级氧化降解水中环境激素
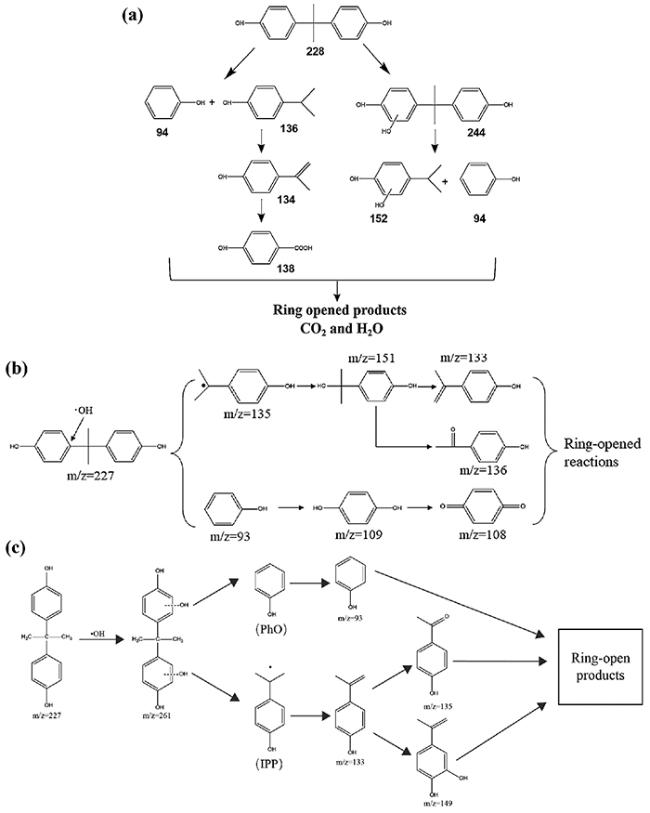
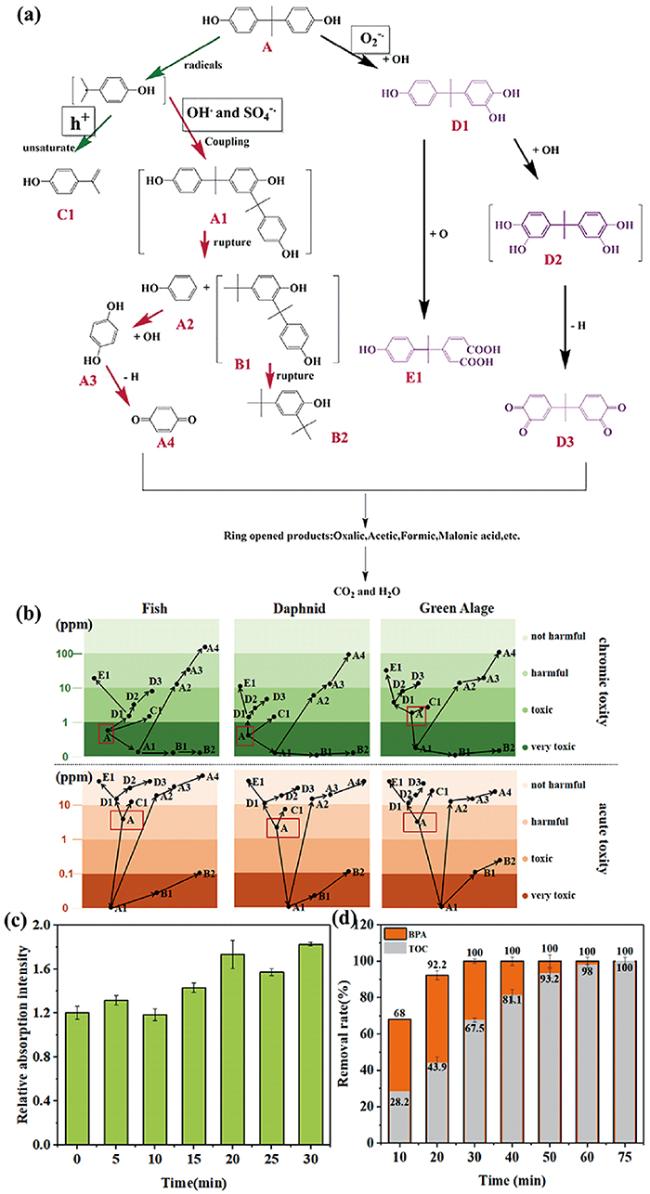
图8 (a) M88/Vis/PS系统中BPA降解的可能转化路径;(b) M88/PS/Vis体系中BPA及其副产物的风险评估;(c) M88/PS/Vis体系对CCK-8的相对吸收强度变化;(d) M88/PS/Vis体系对BPA的矿化效率[63]Fig.8 (a) Proposed transformation pathways for BPA degradation in M88/Vis/PS system;(b) risk assessment of BPA and its by-products via ECOSAR in M88/PS/Vis system;(c) the relative absorption intensity variation of M88/PS/Vis system to CCK-8;(d) the efficiency of BPA mineralization by the M88/PS/Vis system[63] |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
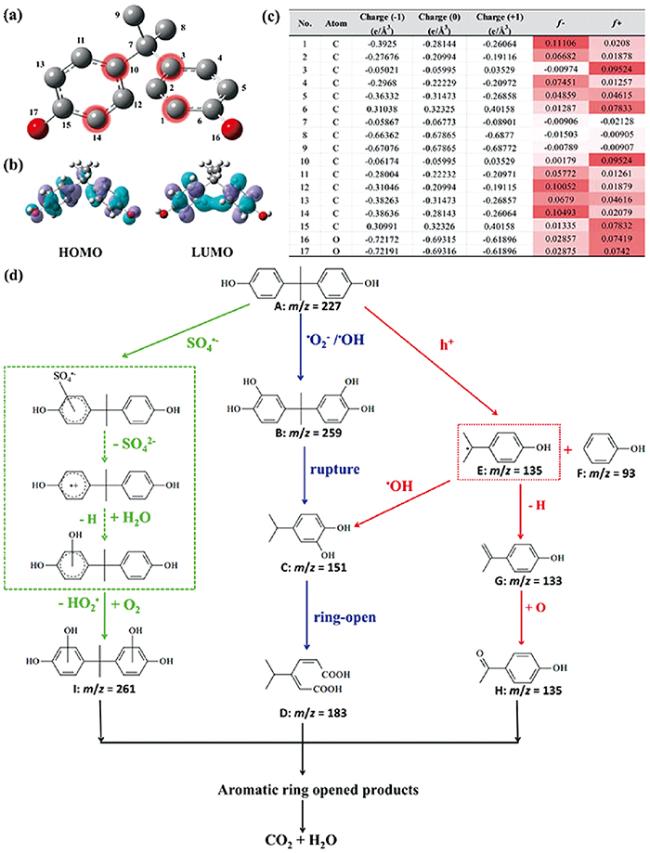
图9 (a) BPA的化学结构;(b) BPA的HOMO和LUMO轨道;(c) BPA的自然种群分析电荷分布与Fukui指数;(d) Bi12O17Cl2/MIL-100(Fe)活化PS降解BPA的可能路径[64]Fig.9 (a) BPA chemical structure;(b) HOMO and LUMO orbitals of BPA;(c) NPA charge distribution and Fukui index of BPA;(d) Proposed pathways of photocatalytic degradation toward BPA over BM200/light/PS system[64] |
3.3 Fe-MOFs及其复合物高级氧化降解水中农药
3.4 Fe-MOFs及其复合物高级氧化同时降解水中多种EOCs
4 Fe-MOFs及其复合物高级氧化降解EOCs的影响因素
4.1 材料本身物理性质的影响
4.2 工艺运行条件的影响
4.2.1 pH的影响
4.2.2 外来离子的影响
4.2.3 其他工艺运行条件的影响
4.3 活性物质的影响
4.3.1 光催化反应中的活性物质
4.3.2 类芬顿反应中的活性物质
4.3.3 活化过硫酸盐反应中的活性物质
 2S +S +· +4H +
2S +S +· +4H +