




1 引言
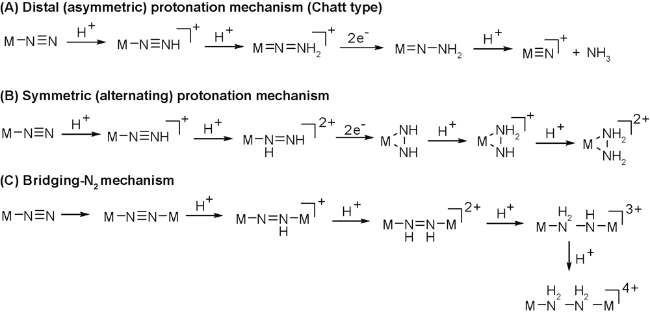
氮气是空气的主要组成部分,是大自然给予我们人类的宝贵资源。作为一个传统的科学难题,如何实现温和条件下氮气的活化及高效转化,使其转变为人类可以使用的资源,是化学家乃至整个科学界的重要研究课题。工业合成氨技术(Haber-Bosch Process)[1]是20世纪人类最伟大的创新之一,它实现了从氮气直接合成氨的工业化生产,为世界经济的发展和人类文明的进步做出了重要贡献。然而,该反应条件非常苛刻,需要高温高压(大约350~500摄氏度,50~200个标准大气压),耗能巨大(据推算每年耗能量约占全球耗能总量的 1%~2%),并且还需要昂贵的催化剂,因此该技术不符合人类可持续发展的要求,也迫使科学家寻找新的方法(反应条件温和、耗能低)[2]来实现氮气的活化和转化。近年来,科学家不断地探索与研究,将氮气转化为含氮化合物的方法也逐渐丰富了起来,大体分为以下几类:(1)人工模拟生物固氮[3]:该方法是模拟植物通过固氮酶将氮气以含氮化合物形式固定下来。人工模拟生物固氮的优点主要有两点:第一是条件温和,第二是产量很高(约占全球每年生物固氮总量的75%)。人工模拟生物固氮也有其缺点:适用范围小,因为具有生物固氮功能的微生物只能与少量的豆科类植物共生,而与我们人类所需要的多数粮食作物并不能共生,因此该方法并不能取代工业合成氨的方法。(2)光催化固氮[4⇓⇓⇓~8]:光催化固氮是利用太阳能将氮气和水合成氨的方法。太阳能作为最清洁的能源之一,利用半导体光催化剂实现氮气转化为氨,无疑是重大的突破。例如:TiO2作为经典的光催化剂及其复合材料被广泛研究并用于光催化固氮[9,10],二元或者多元金属氧化物和硫化物以及各种含碳材料也是近期研究的热点[11⇓⇓⇓⇓⇓⇓~18]。然而,光催化固氮也存在一些显著的缺点:转化效率较低、电子-空穴分离、低量子效率、较差的可见光吸收、选择性差等,主要因为半导体光催化剂表面吸附和催化的活性位点较少。(3)金属配合物催化固氮[19,20]:此方法是先将金属配合物和氮气络合形成络合物,该络合物再进一步通过不同的电子或质子转移路径进行氢化或者质子化[21]得到胺或者其他含氮有机分子,如图1所示。金属配合物具有温和条件下实现氮气活化的巨大潜力,也是未来的研究热点。新型金属配合物的设计与合成是氮气活化与转化的研究重点,金属和配体的高效协同效应将是实现温和条件下催化固氮的有力突破口。
2 含铀金属配合物对氮气的活化
1998年,Scott等[27]首次报道了基于Tren配体三价铀螯合氮气分子的配合物。他们首先使用金属钾还原四价铀的前体化合物1 [U(NN'3)Cl] [NN'3 = N(CH2CH2NSitBuMe2)3]得到混合价态氯桥联的化合物2 [{U(NN'3)}2-μ-Cl][28],再进一步通过高温高真空升华获得重要的三价铀化合物3 [U(NN'3)]。他们发现,3可以在1个标准大气压的氮气氛围中反应生成化合物4 [{U(NN'3)}2(μ2-η2:η2-N2)],颜色也逐渐由紫色变为红色。冷冻-脱气的方法可实现反应的可逆进行。反应过程如图2所示。整个过程可以通过核磁氢谱跟踪的方法来确定反应程度。化合物4 的晶体结构显示氮气分子以侧面配位的方式连接两个铀中心,铀原子分别位于三个氮原子组成的平面之外,约为0.84 Å和0.85 Å。顶端N—U的键长分别为2.555(5) Å和2.601(5) Å,比化合物1中的N—U键(约2.7 Å)和2中的N—U键(2.78(2) Å)都要短,铀与氮气分子中两个氮的键长在2.39~2.44 Å之间。4中的N—N键长约为1.109(7) Å,与氮气中N—N键长(1.0975 Å) [29]的长度基本相同,因而并没有实现真正意义上氮气分子的活化。

金属螯合氮气有端基配位和侧面配位两种方式。在端基配位模式中,两个填充的金属π轨道与氮气形成M→N的反馈键;在侧面配位模式中,仅仅一个金属π轨道和一个δ-轨道可利用[30]。同时,端基配位比侧面配位更有利的原因是π轨道比δ-轨道更有利于配位。
1998年,Cummins等[31]利用异核双金属的策略来实现氮气活化。他们首先用Li(N[R]Ar)(OEt2)为底物和UI3(THF)4反应得到黄色四价铀的化合物5 [UI(NRAr)3],5进一步通过Na/Hg还原得到三价铀化合物6 [U(THF)(NRAr)3]。6与Mo(N[t-Bu]Ph)3在N2氛围下,甲苯为溶剂条件下1∶1反应,几乎定量地生成橘黄色晶体化合物7a(Ph[t-Bu]N)3Mo(μ-N2)U(N[R]Ar)3(图3)。一种合理的推测是可能的中间体(N2)Mo(N[t-Bu]-Ph)3更容易被化合物6捕获,而不是Mo(N[t-Bu]Ph)3本身。采用类似的方法,位阻更大的7b (Ar[Ad]N)3Mo(μ-N2)U(N[R]Ar)3被成功合成(图3)。同时,位阻更大的钼的结构单元促使氮气更倾向于采用端基配位的方式与双金属螯合。7a和7b都通过单晶衍射进一步确定了其结构,结构表明U和Mo都是+4价氧化态,说明发生了两电子的还原。对于双氮复合物而言Mo是更有效的π供体。

2002年,Cloke等[32]采用并环戊二烯配体衍生的三价铀复合物有效地实现了氮气的活化。硅烷化的并环戊二烯双阴离子是一类重要的配体,其螯合的三价铀复合物成功实现了对一氧化碳的键合。受到该工作的启发,他们期待能进一步将该复合物用于氮气的活化。[UI3(THF)4]是合成三价铀前体化合物的重要原料,然而为了合成无强配体配位的并环戊二烯三价铀复合物用于后续小分子的活化,他们采用铀屑和HgI2在封管中320 ℃条件下反应,实现了UI3的合成。UI3分别和等当量KCp*和K2[C8H4{SiiPr3-1,4}2]反应以40%产率得到紫黑色化合物8 [U(η-Cp*)(η-C8H4{SiiPr3-1,4}2)]。将化合物8置于氮气氛围下,核磁氢谱和硅谱都产生新的信号,冷冻之后发现化合物8的信号峰完全消失,产生了新的黑绿色化合物9(图4)。单晶结构显示,化合物9具有双核结构,氮气分子通过侧面配位与两分子化合物8形成复合物。其中N—N键长为1.232(10) Å,与[(TmCp*)2(μ-η2:η2-N2)]中的N—N双键长度(1.259(4) Å)非常接近[33],但显著超过化合物4 [(U{NN'3})2(μ-η2:η2-N2)]中的N—N三键键长(1.109(7) Å)。以上结果也说明化合物3 [U(NN'3)]不能还原氮气的可能原因是:化合物4 [(U{NN'})2(μ-η2:η2-N2)]中配体的空间位阻以及U中心与μ-η2:η2-N2之间的轨道重叠小所导致。虽然化合物3和化合物9中的N—N键的键级存在明显差异,但其U—N键的键长是类似的,这可能是由于两种配体环境中不同的前线轨道几何形状导致的。
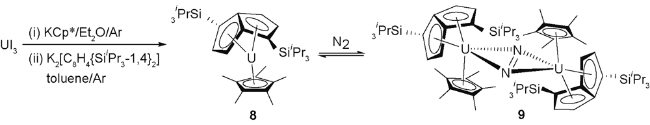
化合物9的形成包含了三价铀到四价铀的转变,并伴随着氮气分子的两电子还原。然而,即使在3.4个标准大气压的氮气氛围下,反应也只能进行到约75%,而9在溶液和固态中都极易失去氮气分子。化合物9失去N2重新生成8的不稳定性可能源于空间比较拥挤,释放氮气后可重新产生平行的三明治结构。
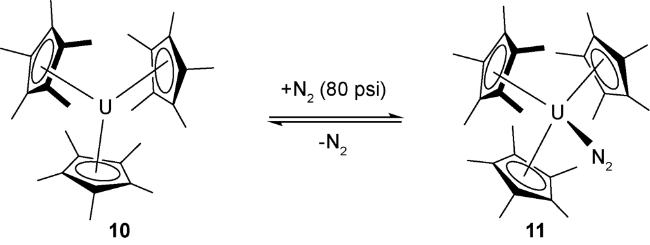
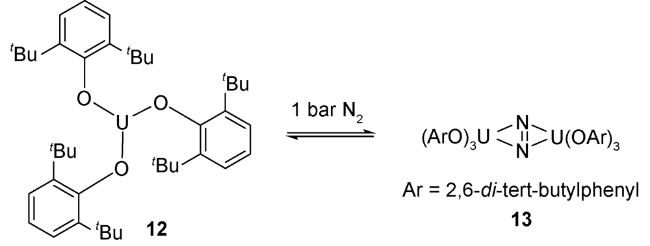
2011年,Arnold等[35]发现利用酚氧络合的三价铀前体化合物12在1个标准大气压条件下也可以实现了对氮气的配位作用形成化合物13。单晶结构表明:氮气分子采用侧面配位的方式与两分子低价铀前体进行络合。其中N—N键长为1.189(18) Å,比化合物11中的N—N键稍长(1.120(14) Å),但是比化合物9中的N—N双键要短(1.232(10)Å),这也说明铀核对氮气具有一定的还原作用。化合物13中铀的π-对称轨道电子反馈到N2的π*-反键轨道,使铀对侧面配位的N2具有更强的活化,形成更加稳定的化合物。
以上内容表明,虽然三价铀作为重要的合成子在氮气的螯合与活化方面展现出一定的潜力,但是氮气转化为胺或者有机胺等重要的化合物仍然没有被报道,主要原因在于以上配合物仅仅能起到配位或者两电子的还原,无法实现氮气的进一步还原和转化。
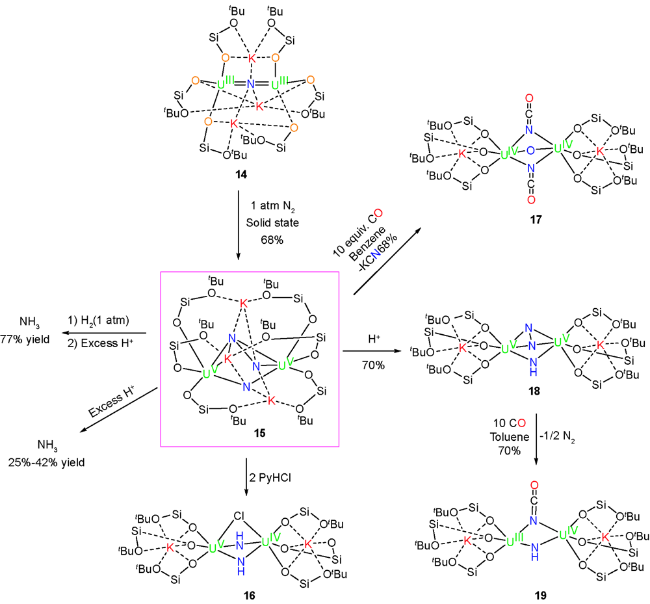
2017年,Mazzanti等[36]采用她们课题组已经发展成熟的大位阻硅醇配体衍生的三价铀复合物,实现了氮气的活化及其高效转化。他们首先使用KC8还原曾经报道的四价铀前体化合物[Cs{[U(OSi(OtBu)3)3]2(μ-N)}]得到重要的三价铀化合物14 [K3{U(OSi(OtBu)3)3}2(μ-N)]。化合物14相比于Cs的类似物而言,具有更高的化学稳定性和更好的结晶性,这也为后续反应的探索提供了先决条件。14的单晶结构显示为氮桥联的两个三价铀螯合的结构单元,同时有三个钾离子进行抗衡。而且硅-氧-钾框架的存在也使柔性的硅醇配体能更好地将两个铀核进行连接,从而能更有效稳定三价铀化合物。在室温条件下,将化合物14以固体形式或者溶于甲苯中,在1个标准大气压的氮气氛围下,可以高效地以68%的产率转化为化合物15。15的单晶结构表明:N—N键长为1.521(18) Å,和NH2-NH2的N—N键长(1.47 Å)非常接近,这表明氮气发生了四电子的还原,铀由三价被氧化为五价,铀的价态也通过电子顺磁共振和变温磁性进行了进一步确认。他们认为柔性的硅-氧-钾骨架在U—N—U核通过弯曲来螯合氮气的过程中起到了关键作用。这也是迄今为止首次发现三价铀复合物可以通过四电子还原来实现氮气活化。
15用盐酸处理可以25%~42%的产率得到氨气;如果先用氢气还原再进一步用酸处理,可以77%产率得到氨气。如果只是氢气还原,则需要两至三周底物才能完全转化。当用两当量PyHCl处理化合物15时,N—N键发生断裂得到化合物16,进一步加大PyHCl至二十当量时,核磁显示氯化铵为唯一的产物。化合物15和一氧化碳反应,可以得到氧代氰酸酯化合物17。17的单晶结构显示双核铀由氧和两个OCN-片段进行桥联。同时,五价铀被还原为四价铀。与此同时,化合物15在2,4,6-三叔丁基苯酚作用下可以得到NH2-和 桥联的双核铀化合物18,18和一氧化碳反应得到NH2-和NCO-桥联的混合价态的U(Ⅲ)和U(Ⅳ)双核铀化合物19,并脱除氮气(图7)。
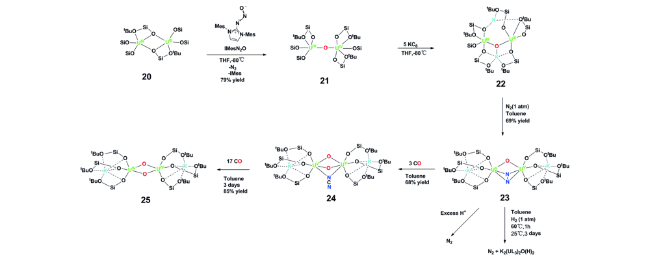
2019年,Mazzanti等[37]报道了一种大位阻硅氧配体支撑的氧桥联双核铀的前体来实现氮气的活化。他们以三价铀化合物20为初始原料,用氮杂环卡宾的一氧化氮复合物处理,得到氧桥联的四价铀化合物21。21被KC8还原得到钾离子抗衡的氧桥联的三价铀化合物22。22可以和氮气发生四电子的氧化还原反应得到化合物23。单晶结构显示N—N键长为1.40(1) Å,和肼的N—N键长(1.47 Å)非常接近,也处于正常的肼类化合物N—N单键键长(1.377~1.548 Å)的范围以内。以上结果有利地说明了氮气发生了四电子的还原得到N—N单键,同时铀从三价被氧化为五价。化合物23和三当量的一氧化碳反应,得到化合物24,单晶结构显示双核铀由两个氧和一个NCN2-单元进行桥联。进一步和17当量的一氧化碳反应,化合物24被进一步还原得到双核铀(Ⅵ)的化合物,同时NCN2-单元被转化为含氮化合物NCNCO进行脱除。然而,与化合物15可以高效转化为含氮化合物不同的是,化合物23不管是用酸处理还是氢气还原,都没有得到相应的含氮化合物,而是以氮气的方式脱除。这种巨大的差异是由碱金属离子的结合方式以及电子结构不同导致的反应性能的显著不同(图8)。
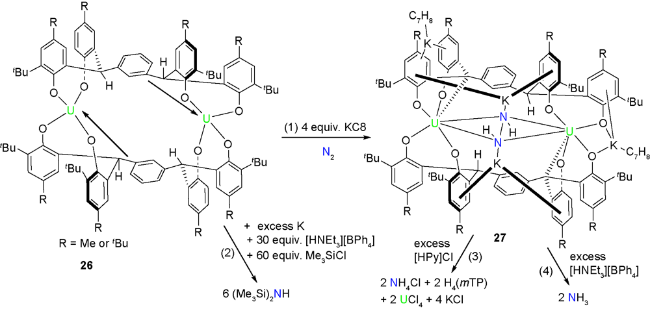
2020年,Arnold等[38]利用刚性骨架的双核铀复合物实现了氮气的还原和功能化,并可以有效地转化为二级硅胺化合物。他们使用间位取代的四芳基酚为配体,得到双核的四价铀化合物26。26的单晶结构可以看出:由于铀的配位不饱和,桥联的芳基和铀之间存在静电相互作用,从而增加其稳定性。26在氮气氛围下KC8还原后,氮气被活化伴随着碳-氢键活化得到多金属螯合的肼类加成物27。27的单晶结构显示三芳基甲烷的碳-氢键发生了活化,氢迁移到氮原子上,并且形成了C—U键。同时,四个钾-芳基的相互作用起到了电荷平衡的作用,而且增强了整体结构稳定性。化合物26和27都可以还原氮气至胺类化合物。26在氮气氛围下用金属钾还原,并进一步采用HNEt3[BPh4]酸化和Me3SiCl硅烷化,得到二硅胺衍生物。27用吡啶盐酸盐处理,分别得到氯化铵、原始配体、四氯化铀和氯化钾;用HNEt3[BPh4]酸化可以得到氨气(图9)。以上结果说明,酸化都可以实现N—N键的断裂,并形成含氮化合物。他们认为双金属双配体的金属大环前体得到的芳基桥联的穴状结构的形成,为氮气的活化提供了关键的作用位点。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
低价铀配合物在氮气活化方面已经展现出巨大的潜力。实现条件温和、低能耗的氮气活化及转化对人类可持续发展具有重要意义,因而应用前景也非常广阔。与过渡金属配合物催化氮气活化相比,低价铀促进的氮气活化具有更加显著的优势,因为铀高度极化,相对论效应导致铀的5f轨道能级更易受晶体场劈裂效应的影响,形成的复合物共价键成分更高;而且三价铀具有很强的还原能力,这些都是低价铀复合物能够高效催化活化氮气的重要原因。当然,铀具有一定放射性,因而存在潜在的危险和危害。因此,同过渡金属相比,含铀化合物在实际操作中的条件要更加苛刻和严格。然而,铀催化的氮气活化也存在一些问题亟待解决:(1)现有的实现氮气活化的低价铀配合物还非常少;(2)已经报道的实现氮气的活化与转化都需要破坏配合物本身,因而催化效率较低。因此,如何实现氮气活化的催化转化与低价铀前体催化剂的循环利用,是未来低价铀催化氮气活化的重要研究内容。当然,这需要合理地设计相关配体以及精准地构筑其配合物,因而还有很长的路要走。但是我们相信,随着研究的深入,真正意义上实现低价铀配合物在氮气活化方面的催化循环指日可待。