



1 引言
自20世纪30年代Reppe首次报道镍催化乙炔、CO以及水合成丙烯酸这一开创性工作以来,过渡金属催化的羰化反应得到了迅速发展[1]。人们陆续实现了以CO为羰源,以烯烃、炔烃、卤代烃和醇等为底物的各类羰化反应,成为生产高附加值大宗化工产品、精细化学品以及医药中间体的重要途径。近年来,随着研究的逐步深入,利用C—H键活化/羰基化反应高效构建羰基化合物的研究成果也不断涌现,甚至成为将羰基引入手性分子的有力手段[2,3]。如Xu课题组通过钯催化的不对称C-H羰基化反应制备得到一系列手性异吲哚啉酮和异喹啉酮[3]。另一方面,为了克服CO高毒性、易燃易爆以及存储运输不便等缺点,发展甲酸、甲醛等新型绿色羰源也成为羰化领域的研究热点[4]。因此,过渡金属催化的羰化反应具有重要的研究意义和广泛的应用前景。
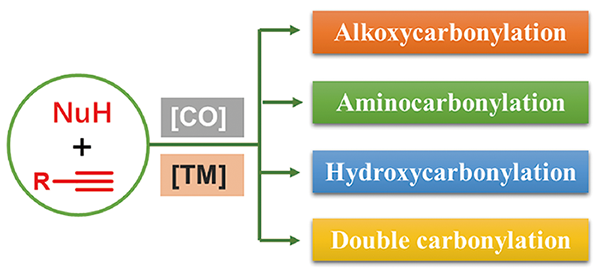
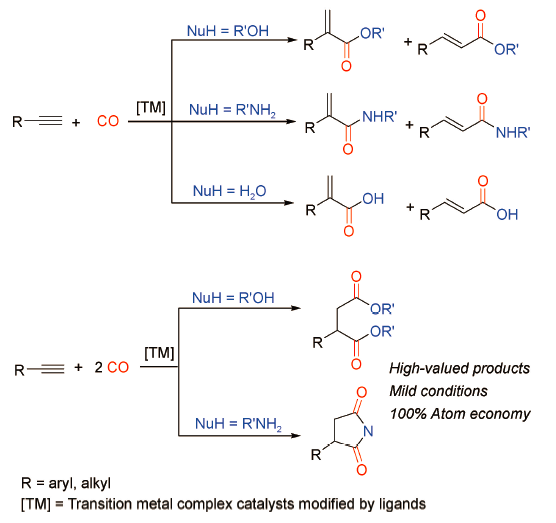
在众多不同原料的羰化反应中,因符合100%原子经济性和绿色化学发展趋势,炔烃的羰化反应日益成为学术界和工业领域的研究热点,并取得了令人瞩目的进展。在过渡金属催化下,炔烃与合成气(CO/H2)为原料通过氢甲酰化反应合成α,β-不饱和醛[5⇓~7],是炔烃羰化研究领域最早被探索的反应过程,也是工业合成醛的有效方法之一。除氢甲酰化以外,炔烃的羰化反应还包括炔烃与CO(或CO替代物)以及亲核试剂NuH(醇、胺、H2O等)作用直接构建α,β-不饱和羧酸及其衍生物等高附加值羰基化合物的反应[8](图式 1 )。根据亲核试剂的不同,羰化反应可以分为以下类型:当亲核试剂为醇时,称为羰化酯化反应(Alkoxycarbonylation);当亲核试剂为有机胺时,称为羰化胺化反应(Aminocarbonylation);当亲核试剂为H2O时,称为氢羧化反应(Hydroxycarbonylation)。此外,炔烃、亲核试剂与两分子CO作用“一锅法”可以构建二羰基化合物,该过程成为炔烃的双羰化反应(Double carbonylation)。
对于羰化反应来说,目前催化剂大多以第Ⅷ族过渡金属为主,如Pd、Fe、Ru、Rh、Ir等[9]。过渡金属催化炔烃的羰化反应体系种类繁多,尽管催化剂、底物、亲核试剂各有不同,但一般公认的反应机理如图式2 所示。
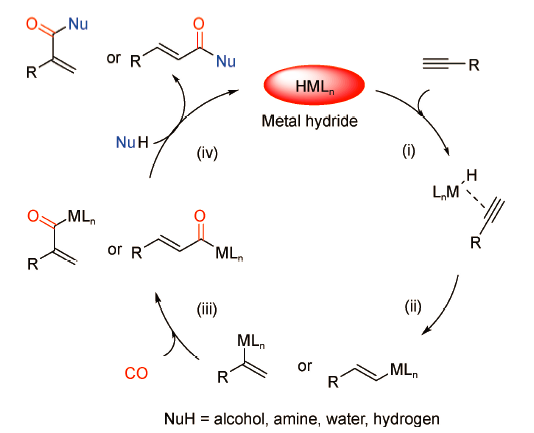
以炔烃为例,整个反应以金属氢化物活性物种的形成为起点:(ⅰ)炔烃C≡C的叁键与金属氢化物的M—H键配位,其配位方向是决定反应区域选择性(直链/支链型产物)的关键因素;(ⅱ)二者进一步加成生成乙烯基金属中间体;(ⅲ)CO插入随后形成酰基金属中间体;(ⅳ)亲核试剂进攻得到目标产物,同时再生金属氢活性物种进入下一轮的催化循环。作为核心驱动力的金属氢化物。在整个反应过程中起到至关重要的作用,其一般通过金属配合物与Brønsted酸助剂(甲烷苯磺酸、对甲苯磺酸等)反应形成。
2 炔烃的羰化酯化反应
由于具有100%原子经济性和操作简便的特点,过渡金属催化的炔烃羰化反应成为构建α,β-不饱和酯类化合物的有效途径之一,广泛应用于香水、调料、医药中间体以及其他化工产品的制备。
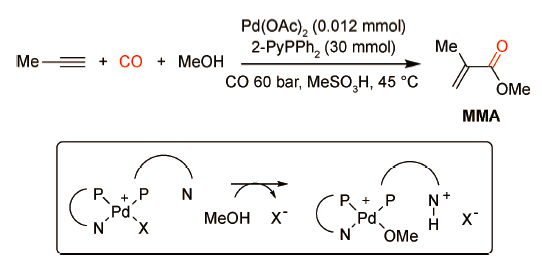
膦配体是过渡金属催化的均相反应中最重要和应用最为广泛的一类配体。通过合理地设计或修饰膦配体可以改变其电子效应和空间效应,从而实现对催化活性以及反应化学/区域选择性的有效调控。因此,开发具有优异催化活性的膦配体是钯催化炔烃羰化酯化反应取得突破性进展的关键,也是科研工作者们普遍关注的焦点。1993年,Drent等[10]开发了一类由Pd(OAc)2,2-PyPPh2以及MeSO3H构建的均相催化体系并将其成功应用于丙炔的羰化酯化反应,实现了丙炔到甲基丙烯酸甲酯(MMA)的高效转化(图式3 )。研究表明,在CO压力60 bar、反应温度45 ℃时,产物甲基丙烯酸甲酯的选择性可达98.9%。作者认为配体2-PyPPh2具有独特的双重功能:一是通过氮、磷原子与钯(Ⅱ)螯合来调控金属中心的电子云密度,稳定Pd金属中心;二是充当“质子信使”的角色,即另一分子配体中未配位的氮原子可作为质子受体捕获、转移氢质子。
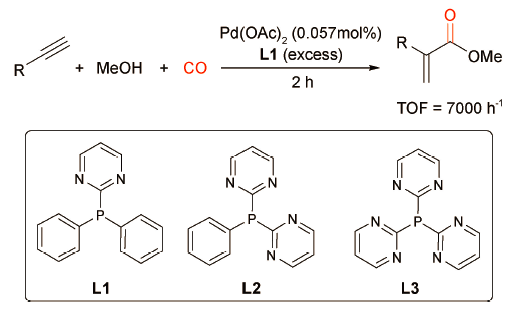
基于这一经典的催化体系,许多科研工作者开始致力于新型含氮膦配体的设计与开发,以进一步提高羰化酯化反应活性。1998年,Reetz课题组[11]合成了一系列含嘧啶基的膦配体L1~L3并将其应用于炔烃的羰化酯化反(图式4 )。研究表明,以Pd(OAc)2为催化剂,L1为配体时,1-己炔的转化率大于97%,TOF可达7000 h-1。当配体换作L2和L3时,1-己炔的转化率依次降低:L1(97%)>L2(70%)>L3(2%)。作者认为,随着膦配体中嘧啶基团的增多,其对P原子的吸电子效应逐渐增强,会打破金属与配体之间的电子平衡,进而抑制炔烃羰化反应的进行。
2001年,Matteoli课题组[12]以二苯基-2-吡啶膦为修饰平台,设计合成了含呋喃环的新型膦配体L4(图式5 )。在CO压力40 atm、反应温度50 ℃时,由Pd(OAc)2/L4/CH3SO3H构建的催化体系可使苯乙炔转化为99.2%产率的支链型酯类产物。该反应体系具有优异的区域性选择性。

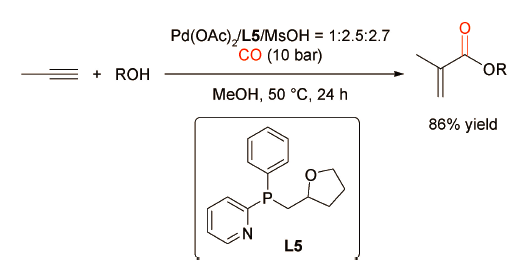
2004年,Green等[13]开发了一种OPN型功能化膦配体L5(图式6 )。L5修饰的钯催化剂对丙炔的羰化酯化反应有较好的催化效果。与2-PyPPh2相比,L5可以更稳定地与Pd金属中心螯合,不需要过多的配体来维持催化活性。该体系唯一的不足是反应时间长,反应速率较低。
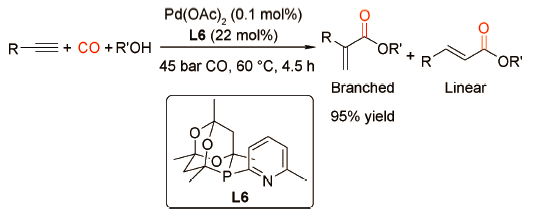
2017年,Shuttleworth课题组[14]构建了一系列含吡啶基的金刚烷膦配体并深入探究吡啶环上的取代基对钯催化炔烃羰化酯化反应的影响(图式7 )。研究表明,当吡啶环6位连有给电子基团—CH3时,配体的催化活性最高;而当连有强吸电子基团—CF3时,催化活性大大降低。使用L6修饰的钯催化剂,在CO 45 bar、60 ℃条件下,苯乙炔可以在4.5 h内完全转化为α,β-不饱和羧酸酯且支链产物选择性高达95%。配体L6同样兼具双齿P-N配体螯合效应和质子转移功能。
2018年,李跃辉课题组[15]以溴代咔唑为原料,通过C-N偶联、C-P偶联方式设计开发了一类基于咔唑刚性骨架的双齿P-N螯合配体L7,发现其在各类脂肪族或芳香族炔烃的羰化酯化反应中均表现出高活性、高选择性(产率45%~96%,支链酯选择性95.0%~99.9%)。X-射线单晶衍射显示,Pd(L)Cl2配合物属于四面体构型,配体L的氮、磷原子与钯金属中心螯合,两个氯离子呈顺式配位。与经典的2-PyPPh2配体相比,这种新开发的双功能配体与钯金属中心显示出更强的配位效应(图式8 )。

2019年,刘晔课题组[16]设计并合成了一种新型的离子型多齿膦配体L8,并成功实现了以水为助剂的炔烃羰化酯化反应(图式 9 )。研究表明,该膦配体存在两种潜在的P-Pd-P螯合方式,可以与Pd生成稳定的络合物;而咪唑鎓的强吸电子效应则使其表现出较强的π-受电子能力。原位红外表征表明,在PdCl2(MeCN)2-L8催化体系中,水作助剂有利于[Pd-H]+活性物种的形成与稳定。

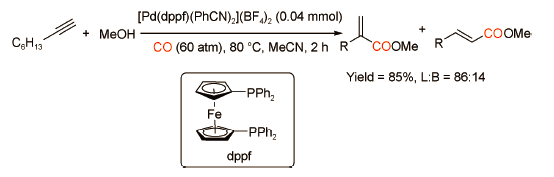
对于炔烃的羰化酯化反应来说,大部分反应体系的产物都以支链型α,β-不饱和酯为主,只有少数催化剂倾向催化炔烃优先生成直链型产物,其中膦配体对区域选择性的调控起到非常关键的作用。Inoue课题组[17]发表了双齿膦配体dppf修饰的钯催化剂催化1-辛炔与CO、MeOH反应生成直链型α,β-不饱和酯的研究(图式10 )。膦配体dppf具有刚性骨架和较大的咬角,而1-辛炔具有大位阻的长链烷基,为了减少空间位阻的影响,Pd—H与炔烃C≡C加成会优先生成直链型的乙烯基钯中间。
2010年,Cole-Hamilton课题组[18]发现以Pd2(dba)3为催化剂,以大位阻的BDTBMB为配体,额外添加MsOH酸助剂,在80 ℃ 、CO 30 bar的反应条件下,苯乙炔主要转化为直链型α,β-不饱和酯——肉桂酸酯,区域选择性达99%(图式11 )。研究机理表明,[Pd-H]+活性物种与炔烃C≡C配位加成时存在空间诱导效应,以直线型乙烯基钯中间体为主,这是反应呈直链型区域选择性的决定性因素。此外,作者利用Pd2(dba)3/BDTPMB/MsOH催化体系实现了对称内炔的羰化酯化反应。

3 炔烃的羰化胺化反应
α,β-不饱和酰胺作为一种重要的化学品在制备天然产物和精细化学品中发挥着至关重要的作用。通过炔烃与有机胺(芳香胺/脂肪胺)或无机铵(NH3/铵盐)可以一步构建α,β-不饱和酰胺。
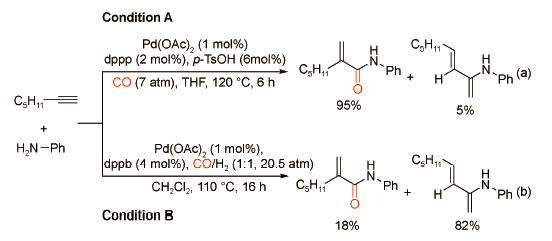
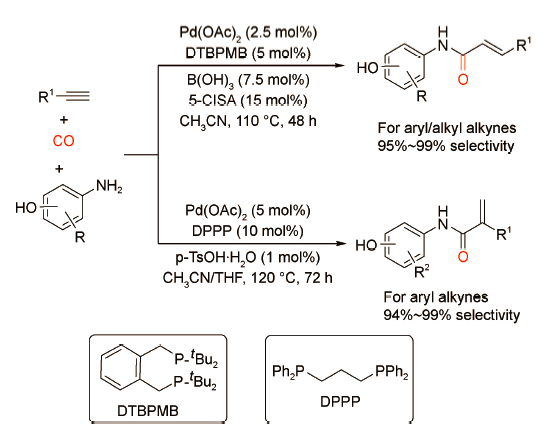
同样地,Alper课题组[21]发现通过简单地改变配体和添加剂,能够以高收率获得支链型或直链型酰胺(图式13 )。作者以对氨基苯酚为亲核试剂,当使用Pd(OAc)2/DTBPMB/B(OH)3/5-ClSA催化体系时,可以获得产率为95%~99%的直链型α,β-不饱和酰胺;当使用Pd(OAc)2/DPPP/p-TsOH催化体系时,可以获得产率为95%~99%的支链型α,β-不饱和酰胺。该催化体系具有优异的化学/区域选择性,广泛的官能团兼容性以及良好的底物普适性,适用于各类炔烃与芳香胺的羰化酯化过程。
与芳香胺相比,具有强碱性的脂肪族胺往往会猝灭氢质子,极大地抑制了钯氢活性物种的形成。因此,以脂肪胺为亲核试剂的羰化胺化反应具有更大的挑战性。
2006年,Alper等[22]发现以离子液体[Bmim]NTf2为溶剂可以成功实现钯催化炔烃与脂肪胺的羰化胺化反应(图式14 )。在温和的反应条件下,无需任何酸助剂,苯乙炔可以高效转化为2-取代丙烯酰胺。离子液体不仅用作反应介质还兼具助剂效应,有利于目标酰胺的生成。作者指出,Tf2N-的弱配位效应赋予[Bmim]NTf2较强的极性,有助于稳定钯金属中心。在循环使用5次之后,该催化体系仍保持优异的催化活性。
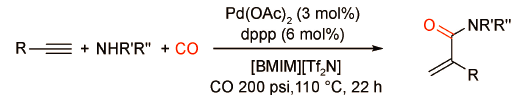
2019年,刘晔课题组[23]在无外加酸助剂条件下,使用dppp修饰的钯催化剂同样实现了炔烃与各类脂肪胺的羰化胺化反应,以高选择性高、产率获得各种支链型酰胺产物(图式15 )。作者指出,在该强碱性条件下[Pd-H]+活性物种难以稳定存在,因此酰胺基钯络合物才是真正的活性中间体。

通常情况下,脂肪族叔胺并非羰化胺化反应的有效胺源。但是,黄汉民课题组[24]通过C—N键活化策略,成功实现了叔胺为胺源的钯催化炔烃羰化胺化反应(图式16 )。研究表明,以5 mol% Pd(dcpf)Cl2为催化剂,以1.0 mmol 过氧化二叔丁基(DTBP)为氧化剂,额外添加10 mol% NH4Cl,在CO压力20 atm、温度120 ℃条件下,苯乙炔可以与三乙胺反应得到77%产率的支链型α,β-不饱和酰胺。该催化体系适用于各类炔烃与脂肪族叔胺的羰化胺化反应。此外,作者将1,1-二苯乙烯作为自由基清除剂引入反应体系,目标产物产率仅下降12%,排除了自由基机理的可能性。

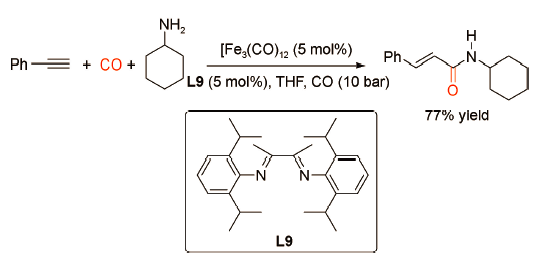
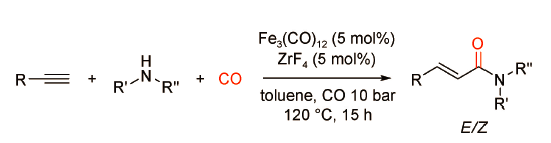
Beller课题组[25]发现相比于贵金属钯催化剂,廉价易得、环境友好的铁催化剂对炔烃与脂肪胺的羰化胺化反应有更加优异的催化效果(图式17 )。以Fe3CO12为催化剂,以L9为配体,在120 ℃、10 bar CO下,苯乙炔与环己胺反应生成直链型的不饱和酰胺,产率为77%。该反应体系具有良好的底物普适性,适用于炔烃与各类脂肪胺的羰化胺化反应,唯一的不足是需要加入过量的胺。李跃辉课题组[26]对铁催化的炔烃羰化胺化反应进行了进一步的改进,发现以氟化锆(ZrF4)为辅助催化剂,更有利于炔烃与脂肪胺向直链型不饱和酰胺的转化(图式18 )。研究表明,ZrF4可以显著影响F…H—N氢键并能够有效活化Fe3(CO)12。
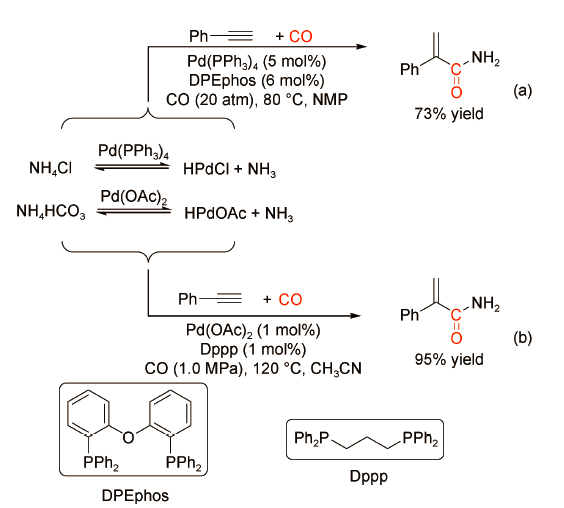
一级酰胺化合物广泛地存在于生物活性分子、药物以及有机功能材料中,具有广泛的应用价值。作为一级酰胺的直接胺源,NH3具有刺激性恶臭气味,在羰化反应中较少使用。无机铵盐因干净清洁、操作简便和存储运输安全等特点而成为氨气的有效替代品。
2018年,黄汉民课题组[27]发现以NH4Cl作为胺源,使用DPEphos膦配体修饰的钯催化剂可以实现各类芳香族和脂肪族炔烃的羰化胺化反应,并且高选择性地获得支链型α,β-不饱和酰胺,产率达到46%~92%(图式19 a)。溶剂NMP可以捕获反应中释放的HCl,促进羰化过程。同年,刘晔课题组[28]利用NH4HCO3作为固体胺源,以Pd(OAc)2与双齿膦配体dppp为催化剂,在反应温度120℃、CO压力1.0 MPa条件下,苯乙炔可以100%的选择性转化为支链型不饱和酰胺,产率高达95%(图式19 -b)。通过对比其他双齿膦配体发现,Dppp的自然咬角为91°,可以保证其与Pd金属中心稳定地配位。此外,NH4HCO3分解释放的H2CO3可以作为一种弱Brønsted酸助剂促进[Pd-H]+活性物种,加快反应进程。
4 炔烃的氢羧化反应
与上述镍催化体系相比,钯催化剂可以在温和条件下表现出良好的催化活性,开发适用于氢羧化反应的高效钯催化体系成为科研工作者的研究热点。2009年,Wang课题组[32]使用Pd(OAc)2/2-PyPPh2/TfOH催化体系实现了乙炔向丙烯酸的高效转化(图式20 )。在最优反应条件下,乙炔的转化率为85%,丙烯酸的选择性为99%,TOF 达到1011 h-1。

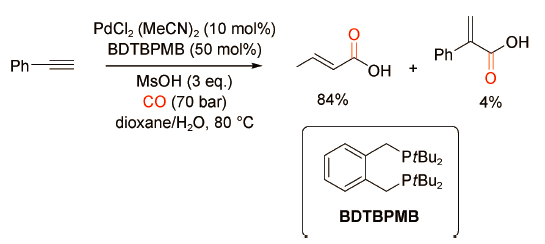
2010年,Cole-Hamilton课题组[18]发现PdCl2(MeCN)2/BDTBPMB/MsOH催化体系可以成功实现苯乙炔的氢羧化反应,该催化体系对直链型α,β-不饱和羧酸具有很高的选择性(图式 21 )。
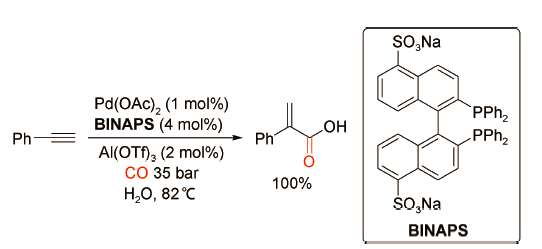
2011年,Williams课题组[33]发现使用Pd(OAc)2、水溶性双齿膦配体BINAPS以及Al(OTf)3构建的催化体系可以在水相中实现苯乙炔的氢羧化反应,目标产物2-苯基丙烯酸的选择性达到100%(图式22 )。然而该催化体系尚无法得到有效的回收利用,这也暴露了其潜在的弱点和局限性。
钯催化炔烃的氢羧化反应通常需要添加Brønsted/Lewis酸助剂以促进[Pd-H]+活性物种的生成。刘晔课题组[34]设计开发了一种兼具P(Ⅲ)配位基团和[P(V)+]Lewis酸性中心的“叔膦-季鏻鎓”水油两亲性三功能配体(图式23 ),该配体在钯催化炔烃的氢羧化反应中发挥了独特的催化效果,在优化的条件下,无需外加酸助剂,便可以得到68%~91%的多种炔烃的氢羧化产物。原位红外表征表明[P(V)+]基团可以活化H2O有利于生成稳定的[Pd-H]+活性物种。此外,以离子液体[Bmim]NTf为溶剂,该催化剂在使用6次后仍具有优异的催化活性,有效解决了催化剂的循环利用问题。
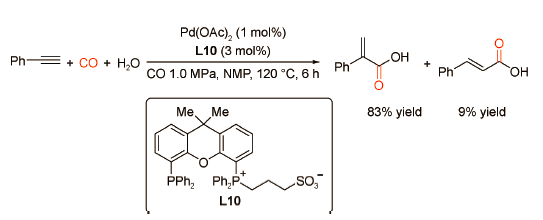
5 炔烃的双羰化反应
炔烃的双羰化反应是羰化反应体系的一个重要研究方向。目前,该反应主要集中于以醇或胺为亲核试剂构建琥珀酸酯或琥珀酰亚胺。
2018年,Beller课题组[35]首次设计并合成了一种含双吡啶取代基的新型双齿膦配体L11,其修饰的钯配合物可显著催化苯乙炔、CO与甲醇发生双羰基化酯化反应直接合成琥珀酸二酯,选择性高达99%(图式24 )。动力学研究和控制实验表明,第一步羰化酯化产物为直链型/支链型α,β-不饱和酯,其在该催化体系下可分别进一步转化为目标二酯,其中支链酯比直链酯在转化过程中更具优势。
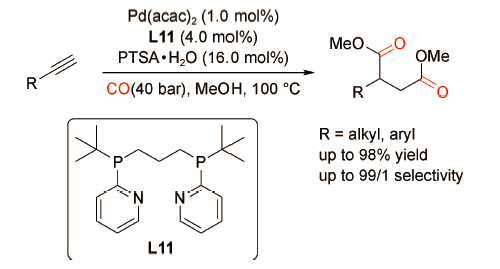
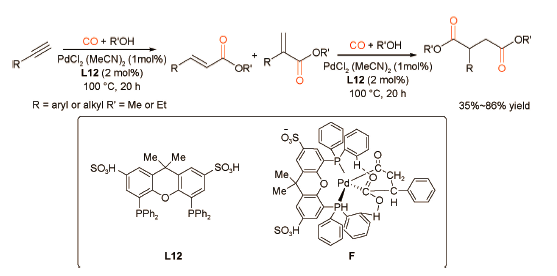
2018年,刘晔课题组[36]制备了一种新型双功能配体L12,该配体中同时含有与Pd金属中心配位的叔膦基团和提供H+的Brønsted酸—SO3H基团。通过构建 PdCl2(MeCN)2-L12催化体系,成功实现了炔烃双羰化酯化反应,高效合成了一系列35%~86%产率的琥珀酸酯衍生物(图式25 )。双功能配体修饰的Pd催化体系的催化活性优于Xantphos 和 MeSO3H 的物理混合体系的催化活性,实现了“1+1>2”的催化效果。通过 DFT 模拟计算发现分子内氢键有利于反应中间体二酰基钯(F)结构的稳定。此外,使用离子液体[Bmim]NTf2作溶剂,可实现该6次循环利用。
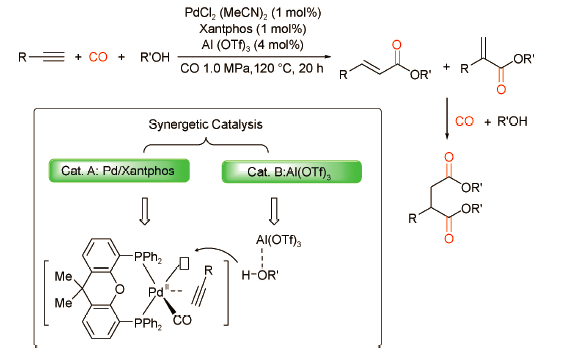
Brønsted酸通常为羰化反应的[Pd-H]+活性物种提供所需氢质子,但过多的Brønsted酸容易导致膦配体的烷基化甚至猝灭活性物种。为有效解决这一问题,刘晔课题组[37]以超强Lewis酸Al(OTf)3代替Brønsted酸构建了PdCl2(MeCN)2/Xantphos-Al(OTf)3双功能催化体系,并通过炔烃的双羰化酯化反应制备了一系列琥珀酸酯衍生物(产率为36%~86%)(图式26 )。在该双功能催化体系中,Pd/Xantphos配合物催化活化底物炔烃和CO,而Al(OTf)3可以通过Lewis酸碱相互作用活化甲醇使其更高效地成为[Pd-H]+活性物种的氢源。
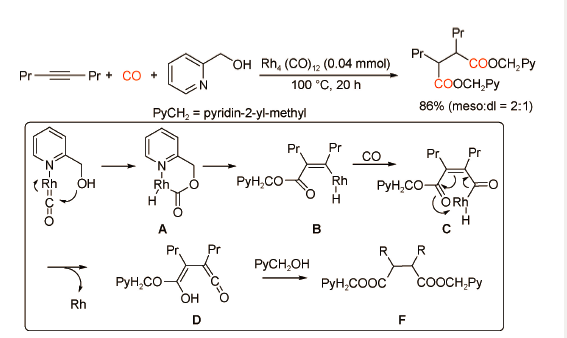
除此钯催化体系之外,Chatani课题组[38]报道了一类特殊的双羰化酯化反应。作者以内炔和2-吡啶甲醇为底物,在Rh4(CO)12催化下合成了一系列1,4-二羧酸酯类化合物(图式27 )。研究发现,与钯催化体系不同,底物2-吡啶甲醇中吡啶N原子可以与铑金属中心配位,形成烯酮中间体,进而推动后续羰化过程。
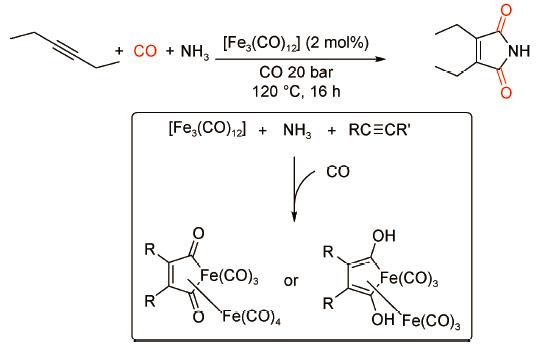
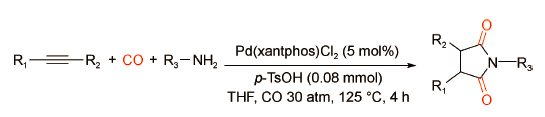
2009年,Beller课题组[41]首次报道了由廉价金属Fe催化的3-己炔的双羰化胺化反应(图式 28 )。研究表明,以2 mol%的[Fe3(CO)12]为催化剂,在反应温度120℃、CO压力20 bar条件下,3-己炔与氨气反应可以得到93%收率的3,4-二乙基琥珀酰亚胺。与传统金属氢化物机理不同,环铁络合物是该催化体系的关键中间体。该体系的亮点在于以廉价金属铁为催化剂,摆脱了对传统贵金属的依赖,并且无需外加配体,经济高效;但是底物仅局限于氨气,限制了产物的多样性发展。2015年,Dyson课题组[42]利用Pd(xantphos)Cl2/p-TsOH催化体系实现了炔烃与各类有机胺的双羰化胺化反应。与脂肪胺相比,碱性更弱的芳香胺可以更高效地转化为酰亚胺产物(图式 29 )。
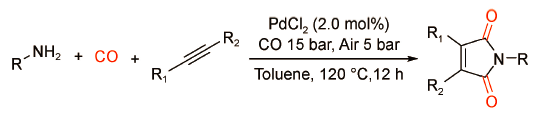
以上催化体系虽然取得了不错的催化效果,但是存在一个共性问题,即它们只适用于内炔,尚无法实现端炔的双羰化胺化反应。2018年,Beller课题组[43]以空气为绿色氧化剂,通过钯催化氧化羰化反应制备了一系列酰亚胺化合物(图式 30 )。该催化体系不需要昂贵的配体或添加剂,经济高效;具有良好的官能团兼容性、广泛的底物普适性以及优异的区域选择性,适用于各类内炔或端炔与有机胺的双羰化胺化反应。
6 炔烃羰化反应中的羰源
6.1 以甲酸酯为羰源
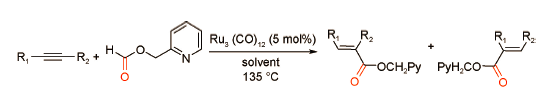
甲酸酯类化合物是炔烃羰化酯化反应的CO常用替代物,具有稳定高效的特点。2003年,Chang课题组[48]以2-吡啶基甲基甲酸酯为CO替代品通过螯合辅助策略实现了钌催化的炔烃羰化酯化反应(图式 31 )。在Ru3(CO)12催化作用下,该反应体系可得到42%~89%产率的α,β-不饱和酯且具有良好的区域选择性。作者认为,底物2-吡啶基-甲基甲酸酯不仅用作替代羰源,而且兼具N配体的配位作用,其N原子可以与Ru金属中心配位形成稳定的六元环过渡态配合物,进而有效抑制酰基氢化物中间体的脱羰过程。
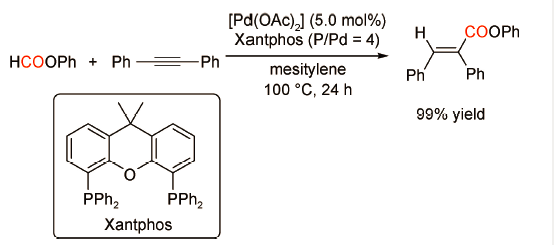
2011年,Tsuji课题组[49]报道了以甲酸苯酯为羰源的炔烃羰化酯化反应(图式 32 )。在优化的反应条件(Pd(OAc)2 5 mol%,Xantphos 10 mol%,100 ℃,24 h)下,二苯乙炔可实现向目标产物E-不饱和酯的高效转化,产率高达99%。机理研究表明,在催化剂作用下甲酸苯酯原位分解生成CO与苯酚,炔烃继而发生后续的羰化酯化反应。值得注意的是,双齿膦配体Xantphos在甲酸苯酯的转化和炔烃的羰化酯化过程中均表现出优异的催化性能。此外,甲酸苯酯的使用对反应的顺利进行至关重要,烷基甲酸酯类产品譬如甲酸丁酯、甲酸-2-苯乙酯、甲酸苄酯等在该催化体系下均不发生转化。
6.2 以甲酸为羰源
作为一种廉价易得、环境友好的可再生C1资源,甲酸既可以通过生物质氧化得到,又可以由二氧化碳催化加氢制备,被视作理想、绿色环保的替代羰源,被广泛应用于炔烃的氢羧化反应中。
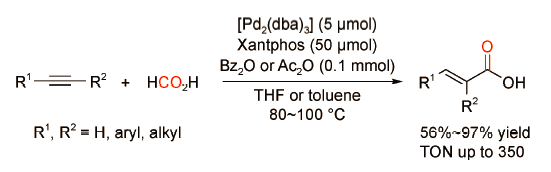
2015年,周其林课题组[50]首次报道了以甲酸为羰源的炔烃氢羧化反应(图式 33 )。作者以[Pd2(dba)3]·CHCl3和双齿膦配体Xantphos为催化剂,额外添加催化量的苯甲酸酐,在反应温度为100 ℃条件下反应转化数TON可达到350。此外,该催化体系也适用于其他端炔或内炔的高效氢羧化反应,产率可达56%~97%。
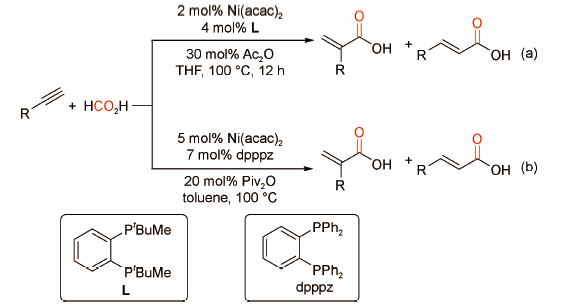
6.3 以金属羰基化合物为羰源
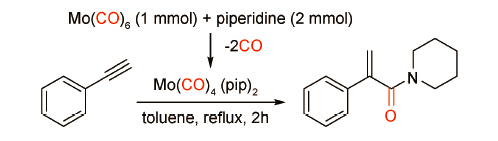
六羰基钼(Mo(CO)6)是一种使用方便的固体羰源。2015年,Babu课题组[53]以苯乙炔和哌啶为模板底物,以Mo(CO)6为催化剂和羰源,在异丁醛辅助催化下,可以得到产率为83%的支链型酰胺(图式 35 )。作者指出,Mo(CO)6与哌啶会形成一种Mo(CO)4(pip)2络合物,既是催化剂活性中间体又可以提供反应所需CO。该催化体系底物具有广泛的底物普适性,适用于各类端炔与二级脂肪胺的羰化胺化反应。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
7 结论与展望
过渡金属Pd、Fe、Ru催化的炔烃羰化反应已成为制备羧酸及其衍生物的重要绿色途径之一。迄今为止,科研工作者已发展了众多具有优异催化活性的催化体系,高效实现了炔烃的羰化酯化反应、氢羧化反应、羰化胺化反应、双羰化反应等合成羰基化合物的过程。
目前炔烃的羰化反应主要以CO为羰源,针对CO高毒易燃、高压操作不便等问题,采用可再生、低毒的液态或固态CO替代品进行羰化反应,也成为近年来炔烃羰化反应的重要研究方向。目前,可适用于炔烃羰化反应的可替代CO的羰源包括甲酸、甲酸酯、金属羰基化合物等。但这些CO替代物的适用范围比较单一,且对催化体系要求较苛刻。因此,廉价、丰富的CO气体仍将是羰化反应广泛采用的羰源,尤其是在羰化反应工业化应用过程,CO作为羰源让具有不可替代的地位。此外,以CO2为羰源制取羰基化合物符合绿色化学发展趋势,但是CO2在炔烃羰化反应领域的应用仍然面临巨大挑战。因此,在这一研究方面有很大的探索空间。