




1 引言
FeOCl是一种典型的层状材料,早在20世纪30年代,德国科学家Schäfer就在部分热分解的FeCl3·6H2O中发现了FeOCl[6]。随后在海洋自然环境中人们也发现了FeOCl的存在,如在海底打捞的文物表面常常伴生FeOCl,对其的妥善移除对海底文物出水和防腐工作至关重要。20世纪70年代超分子化学的兴起,FeOCl作为一类有趣的无机主体,在有机-无机插层化学领域被广泛研究,形成了FeOCl材料的第一次研究高潮(图1a)[7]。但是在此期间,除了少量研究以FeOCl作为锂离子电池的正极材料以外,鲜有FeOCl应用相关的研究[8]。这种状况的改变起于2013年,本课题组发现FeOCl是一类优异的固体Fenton催化剂,其可以以高于其他铁(氢)氧化物1~3个数量级的能力活化H2O2生成羟基自由基(HO·),实现水中污染物的降解[9]。自此,FeOCl被作为水处理催化剂进行了广泛的研究,研究者围绕其结构改性(形貌优化、剥离、插层等)、工程化(负载化)、反应体系强化(光辅助、超声辅助等)进行了大量的工作。另外,FeOCl在其他氧化体系(如苯羟基化)中也展现了其优异的催化活性[10]。除了催化应用以外,FeOCl在可充放电氯离子电池、超级电容器电极材料的研究中也取得了一定的进展[11,12]。FeOCl还可充当前驱体、模板或界面交联层,用于其他纳米材料复合结构的构筑[13,14]。
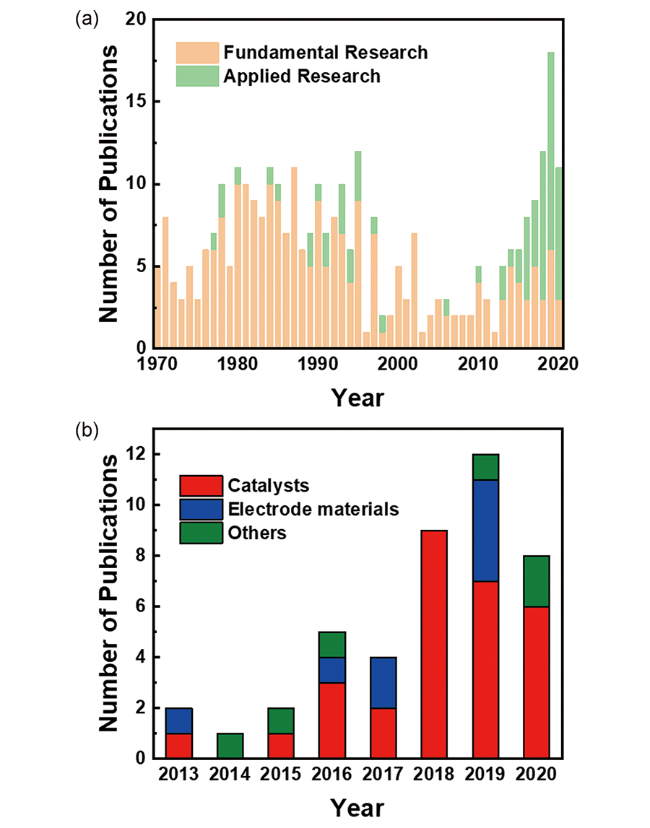
图1 自1970年以来FeOCl的研究论文发表数(a)和自2013年以来FeOCl应用性研究的论文发表数(b)(基于Scifinder和Web of Science检索结果,2020年数据截止至3月)Fig. 1 The number of papers about FeOCl since 1970(a) and the number of papers dealing with the application of FeOCl since 2013(b)(The results are based on Scifinder and Web of Science, data as of March 2020) |
材料科学家们围绕经典的层状材料,如MoS2、石墨烯,已经积累了大量的研究经验,对于其可控制备、结构改性和相关的应用已经有了较为深入的认识。与这些经典体系相比,FeOCl中主体Fe元素环境丰度大(地壳含量排名第四),价态变化灵活,且无毒无害,其他层状材料难以兼具这些优势。虽然FeOCl已经展现了巨大的研究潜力,但是我们对于其性质认识仍较为浅显,对于其现有研究也缺乏系统的梳理,这限制了FeOCl材料的进一步开发。对于FeOCl结构的全面认识和现有研究的总结是推动其应用研究进一步开展的必要前提,在本综述中我们首先对FeOCl结构及以插层反应为代表的结构改性研究进行了总结,这些研究主要集中在1970~2000年。另外,我们对2013年以来关于FeOCl的各种应用研究进行了总结,并从FeOCl结构设计的角度对其进一步的应用开发进行了展望。
2 FeOCl的制备与结构
2.1 FeOCl的制备
2.1.1 FeOCl块体材料
而化学气相迁移法以无水氯化铁和氧化铁(赤铁矿,α-Fe2O3)为前驱体,需要在无水氯化铁过量的情况下将二者在无水无氧条件下充分混合,将混合后的固体焙烧,在焙烧过程中无水氯化铁会发生气化,并在氧化铁的气-固界面发生拓扑转换,生成FeOCl3,再经过FeOCl3的分解和结构重排生成FeOCl。由于反应体系的不均匀性,在FeOCl3结构重排的过程中也可能释放氯气(Cl2)。其反应方程式可以用(3)和(4)表示:
这两种方法制备得到的FeOCl形貌结构存在较大的差异,部分热解法制备的FeOCl尺寸较小,形貌较不规则,而化学气相迁移法制备的FeOCl结构规整、结晶度较高。因此后者虽然过程繁琐,但是在插层化学以及一些应用研究中受到了更多的关注。我们一般认为部分热解法制备的FeOCl稳定性较差,但是近期在一些应用研究中通过一定的制备条件控制(如焙烧温度、时间、气氛等)也制备到了稳定性较好的FeOCl材料[17]。关于FeOCl块体材料制备中涉及的诸多机理性问题(如相转化行为、晶体的生长行为等)目前还不甚了解,经验性的制备条件变化还是制备方法优化的主要手段。此外,一些其他的制备方法,如溶液浸渍-焙烧、液体脉冲激光烧蚀(LAL)等也在近期的研究中被开发,我们将在应用研究中进行介绍。
2.1.2 FeOCl二维材料
作为一类典型的层状材料,从理论上讲FeOCl也可以像其他层状材料(如石墨、MoS2)一样通过剥离或气相生长等方法制备,但是由于FeOCl材料的研究尚处起步阶段,目前仅有少量研究涉及了FeOCl二维材料的制备及性能。在FeOCl的气相生长(化学气相迁移)制备过程中,前驱体的混合比率、升温速率、反应器容积等因素会影响FeOCl片层的尺寸和厚度。本课题组曾通过改变FeOCl制备过程中的反应器容积来尝试调控FeCl3的气化分压,发现FeCl3气相分压的增加(容积尺寸降低)会抑制FeOCl(010)晶面的增长,但是未能实现二维厚度FeOCl纳米片的制备[18]。尽管目前二维FeOCl纳米片的直接气相生长制备尚未有成功先例,但是通过液相剥离的方法由FeOCl块体材料制备二维FeOCl纳米片的研究已有报道。和传统范德华层状材料的剥离方法类似,Zhang等和Schoop等[17,98]分别利用超声剥离和正丁基锂插层剥离的方法成功实现了少层FeOCl纳米片的制备,制备得到的材料在H2O2活化行为及电磁性质上较块体FeOCl都有了显著的变化,对此我们会在相应的应用研究部分进一步介绍。总之,FeOCl虽然是一类新型的层状材料,但是其具有与传统范德华层状材料类似的性质,可以借鉴其他材料的开发思路实现FeOCl二维材料的设计与制备。
2.2 FeOCl的结构
Goldsztaub[19]曾于20世纪30年代就对FeOCl的晶体结构进行了测定,但由于材料结晶度较差,所得到的晶胞参数存在一定误差。此后,在穆斯堡尔谱(穆谱)的发展过程中,由于FeOCl具有较强的核四级相互作用,其被作为确定穆谱57Fe的核四级矩数值的绝佳材料,对其晶体学参数进行了一定的研究[20,21]。1970年,Lind[22]利用单晶X射线衍射对FeOCl的晶体结构进行了重新测定,得到了更为精确的结构信息,并一直沿用至今。在后续插层化学的研究中,人们在研究插层产物的过程中也会同时测定原始FeOCl的结构参数,获得的数字虽有一定差异,但差别往往并不大。FeOCl晶体属于正交晶系,空间群为Pmnm(D2h13),晶胞参数为a=(3.780±0.005)Å、b=(7.917±0.005)Å、c=(3.302±0.005)Å,其典型结构见图2。在FeOCl中,每个Fe原子与4个O原子、2个Cl原子配位,呈顺式cis-[FeO4Cl2]八面体,八面体之间共享O—O或O—Cl共边连接形成双层,双层外再密堆一层Cl原子,由于这层Cl原子的范德华作用,使得FeOCl具有层状结构。

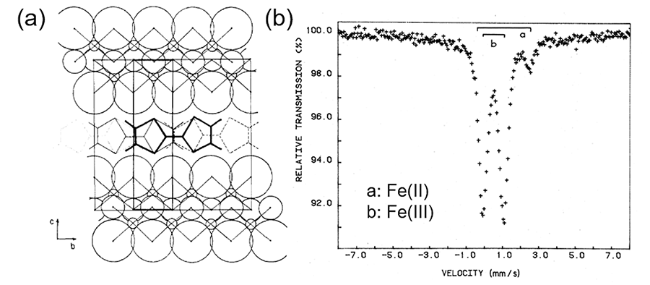
Fe—O、Fe—Cl、Fe—Fe之间的键长往往最能反映Fe所处的化学环境,这也是对理解FeOCl的反应性最为主要的结构参数。FeOCl中Fe—O键长分别为1.964和2.100 Å,与γ-FeOOH、α-Fe2O3中的Fe—O键长基本类似,比石榴石和钙钛矿结构中的Fe—O键短一些。Fe—Cl键长2.368 Å与FeCl3的Fe—Cl离子键键长近似、比[FeCl2(H2O)4]+中Fe—Cl键长很多,显示其离子性可能更强。由于z方向上O和Cl的电负性、原子作用半径和成键能力的一系列差异,导致空间延展的共边八面体层较γ-FeOOH、α-FeOOH、α-Fe2O3层发生了扭曲,这反映在(001)晶面两侧的O-Fe-Cl和O-Fe-O的键角分别为172.39°和148.48°,而上述铁氧化物类似拓扑方向上的键角是等值的,都为160°左右。在沿(001)晶面上的Fe-O之间的键角则与α-Fe2O3中没有太显著的区别。外延的Cl—Cl之间间距为3.68 Å,基本上是两倍于Cl的范德华作用半径,形成典型的范德华层。除了X射线衍射以外,显微成像技术也在FeOCl晶体结构的认识上发挥了很大的作用。1995年,Pieczko和Breen第一次利用原子力显微镜对FeOCl进行了研究[23],表面像显示其在成像区域内(10 μm×10 μm)呈现出光滑表面,而原子分辨率像显示其端位Cl原子具有明显的范德华层结构,对其表面原子周期性结构的测量也与XRD晶体学数据基本一致。另外,在FeOCl的部分区域其表面具有明显的表面缺陷,高度差在0.70 nm和1.31 nm,这可能对应于FeOCl的层高度,但是也观察到一些几微米深的孔洞,这可能是由于FeOCl在空气气氛中反应或受电子束辐照损伤所致。
自20世纪70年代以来,人们利用穆斯堡尔谱、理论计算等技术对FeOCl电子性质和磁性质等进行了大量的研究。根据穆斯堡尔谱的研究,在300 K时FeOCl中Fe原子的特征信号与非对称高自旋Fe3+基本相同;在低温时,FeOCl的有序排列呈现出反铁磁性,但关于有序化温度值的报道不同文献不尽相同,有的报道(68±2)K[24],也有的报道为(85±1)K[25]或(90±1)K[26],除了早期测量技术对结果的影响以外,这些实验值的差异可能反应了FeOCl结晶度、晶面取向、杂质组分以及空气稳定性等因素对其结构和物性的影响。FeOCl对水的敏感性也可进一步体现在其Mossbauer晶格温度的差异[26],采用对前驱体都严格地控水控湿的制备工艺获得的FeOCl样品Θm为(327±6)K,而在不采用这些获得的样品的则为Θm为(267±5)K。室温下FeOCl的摩尔磁化率值χm为3.86×10-3或 3.26×10-3,随着温度降低χm的值在逐渐下降,但是在经过有序化温度点时并未发现明显的磁异常现象。反铁磁结构是非线性的,存在两个不等性的Fe3+位[27]。FeOCl中Fe的d轨道裂分为eg和半填充的t2g;O的p轨道填充影响邻近两个eg和t2g的耦合,从而影响顺铁磁的直接交换耦合和反铁磁的超交换耦合,耦合强度直接受到结构上Fe-O-Fe键长和键角的影响。FeOCl中的电子自旋方向分别与其b轴和c轴平行,由于与范德华层中反磁超交换耦合存在竞争而产生依时的自旋弛豫[25]。FeOCl呈现出半导体性质,其电阻率约为 107 Ω,电子供体和Lewis碱的插层都会不同程度地降低其阻抗 [7]。采用Mossbauer谱、X射线吸收等方法结合价带模型(band model)、团簇模型(cluster model)、电子转移模型(charge transfer model)等[28,29]的计算可进一步揭示FeOCl的电子结构以及FeOCl与插层客体之间的电子转移[30,31]和电子跃迁过程[32]。其态密度(DOS)、局域态密度(PDOS)信息表明FeOCl费米能级附近的电子密度主要由Fe-3d轨道贡献,因此在发生电子转移行为(如插层反应)时主要对Fe原子产生影响;而电子轨道重叠布居(COOP)图则表明位于费米能级附近的分子轨道是Fe、Cl的弱成键轨道,而费米能级以上的轨道是Fe和O的反键轨道,其在插层过程中容易被客体转移的电子占据[33]。
3 FeOCl插层改性
3.1 插层机理
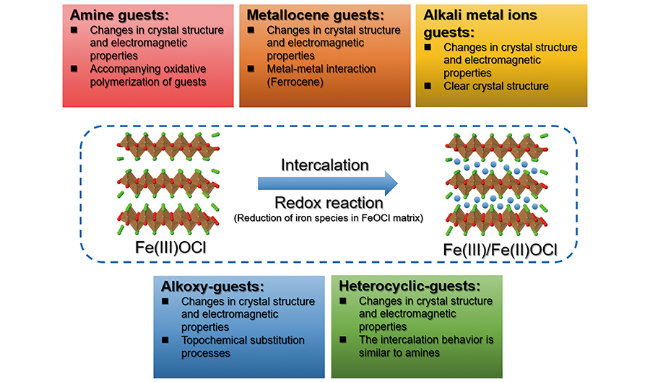
层状化合物插层机理的研究始于经典的层状过渡金属氧(族)化合物(如MoS2),由于大多数插层客体都存在孤对电子,即具有Lewis碱特征,早期的研究认为插层反应是由于主体-客体之间的Lewis酸-碱相互作用导致的[29]。在插层结构中,客体物种的孤对电子与主体金属原子dz2空轨道之间发生重叠,形成新的化学键。然而,随着研究的深入,特别是NMR、中子衍射和穆谱等表征技术的发展,表明在插层过程中客体物种的孤对电子是沿层方向平行排布,不能与主体金属dz2轨道成键,且在插层化合物中观察到了主体结构金属的还原[32,34⇓ ~36]。因此,研究者们逐渐认为主体和客体物种之间的氧化还原反应才是插层的诱因 。新的电荷转移模型被提出来解释插层过程,即客体物种价带上的离域电子诱导主体骨架物种发生还原,再与客体阳离子或质子化物种通过电荷相互作用形成稳定的插层化合物。此后针对FeOCl插层过程的诸多研究及表征也证实了FeOCl的插层和其他层状化合物一样遵循基于电荷转移的氧化还原机理(图3),且FeOCl的氧化性更强,插层过程更为激烈、迅速[29]。
3.2 FeOCl的插层结构演化
对于FeOCl而言,由于插层过程伴随的客体物种的插入和主体(一般是骨架Fe原子)还原,FeOCl插层化合物较原始FeOCl在晶体结构、电子性质和磁性质上往往都会发生巨大的变化。晶体学衍射、穆谱、同步辐射X射线吸收谱和理论计算研究可得到FeOCl插层行为和FeOCl插层化合物性质变化的大量信息,使人们对于这一材料的认识深度大大增加。
由于插层客体往往具有较大的尺寸,对FeOCl晶体结构最主要的影响就是引起垂直于层方向(b轴)上的层间膨胀。在现有的文献报道中,除了Li离子以外,几乎所有客体物种的插层都会引起FeOCl层间距的增加。这种层间距的膨胀可以直接通过XRD进行观察,在一些定域电子衍射(ED)结果中也观察到了这种现象[37]。除了层间距的变化,客体插层还会引起FeOCl层内晶胞参数(a、c)的变化,这主要是由于Fe(Ⅲ)的还原引起配位场环境进一步畸变和层间客体物种与端位Cl原子的配位所引起的[37]。另外,由于FeOCl垂直于层方向n滑移面的存在,插层过程可能会引起FeOCl层结构的滑动,破坏其结构对称性,造成空间群的改变。如四硫富瓦烯插层的FeOCl呈现出体心对称性,空间群变为Immm[36,38];甲醇盐插层的FeOCl空间群变为Amma[39]等等。但需要注意的是,由于X射线对轻元素衍射能力较差、插层后晶体劣化等因素,早期(21世纪之前)对FeOCl插层化合物空间群的准确指认存在较大的困难。近年来,随着同步辐射X射线光源和散裂中子源等大科学装置的兴建,同步辐射X射线衍射、中子衍射等技术在材料表征中的作用日益增加,它们的空间分辨率较传统的衍射技术大大增强,可以获得高强度、高质量的衍射数据以供结构解析。另外,一些测试的附属装置(如全流程惰性气氛制样)也日臻完善。这些技术在很大程度上提升了对FeOCl插层化合物结构解析的成功率和可靠性,极大地推动了对其晶体学结构的认识。
除了FeOCl插层化合物的晶体学演化过程,其电子性质和磁性质的变化也得到了广泛的研究。尽管客体物种存在差异,但穆谱研究的结果都表明FeOCl的插层过程伴随着Fe(Ⅲ)的还原,Fe(Ⅱ)比率在0.10到0.13之间,且与客体物种的种类几乎没有明显的关系。变温实验的结果表明在低温时FeOCl插层化合物呈现出磁有序性,Fe(Ⅲ)物种的同质异能位移与纯相FeOCl一致,同时可以观察到明显的Fe(Ⅱ)共振的四级分裂。随着温度的升高,Fe(Ⅱ)比例逐渐减小,Fe(Ⅲ)共振的四级分裂减弱。根据变温过程中Fe(Ⅲ)、Fe(Ⅱ)穆谱特征参数的变化情况,可以认为Fe位点周围的平均电子环境存在快速的电子交换,这一过程通过电子在不同价态Fe位点之间的跃迁实现,根据变温实验的结果电子跃迁的活化能约为5.0±0.9 kJ/mol[20,29,32]。虽然穆谱在对FeOCl插层电荷转移过程的研究中做出了很大的贡献,但是由于穆斯堡尔效应自身时间尺度(~10-8s)的限制,难以对电子转移过程进行精确描述和定量。近年有研究利用同步辐射X射线吸收谱和发射谱对FeOCl电荷转移过程进行了探究,其可以实现~10-16s时间尺度上对电子跃迁行为的表征。利用此技术对Na、聚苯胺插层的FeOCl的研究发现其Fe(Ⅱ)比率约为0.25,远高于穆谱表征的结果[31]。
3.3 FeOCl插层化合物
目前,多种化合物(如胺类、茂金属、碱金属阳离子、烷氧基化合物(醇盐类)及其他杂环化合物等)都被作为客体物种用于FeOCl的插层研究,它们具有不同的结构和化学性质,因此插层过程和形成的插层化合物性质也有很大的差异,下面将逐一对其进行介绍。
3.3.1 胺类插层化合物
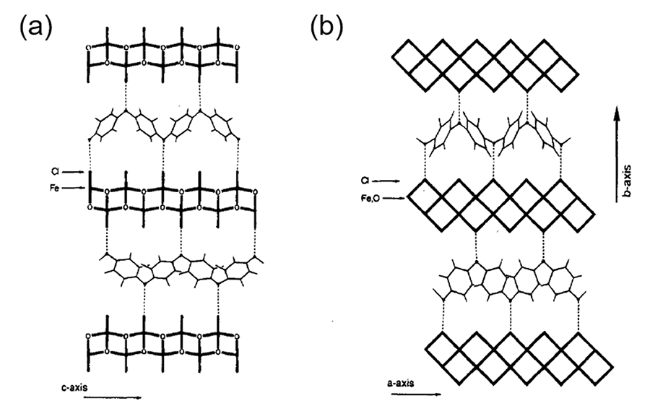
胺类化合物,如苯胺、吡啶、吡咯等,是一类最常见的Lewis碱,对其在FeOCl的插层反应中进行了大量的研究,是目前研究最为广泛也是最为重要的一类FeOCl插层客体。Maeda等[40]在乙醇中将苯胺等一系列胺类化合物与FeOCl共同浸泡,制备了多种胺类插层FeOCl,胺类物种在层间作为电子供体会发生氧化,形成聚合物(如聚苯胺(PANI))。他们发现客体物种的酸性对插层化合物的合成有明显的影响,只有酸解离常数(pKa)大于5的胺类才能有效插入FeOCl层间。Wu等[41]详细研究了苯胺与FeOCl的插层聚合反应,对插层过程和产物在空气氛围中的稳定性进行了详细的表征。穆谱显示PANI插层FeOCl的Fe(Ⅱ)/Fe(Ⅲ)比例约为1/9。X射线单晶衍射的结果显示苯胺的插入诱导FeOCl生成2a×2c的超结构。层间PANI的重均分子量(Mw)约为6100,数均分子量(Mn)约为3500,远低于常规开放式聚合方法合成的PANI(Mw约为69 000,Mn约为7700),其在层间具有较短的链长和较窄的长度分布,变温氢谱NMR显示PANI的苯环旋转在层间明显受限。基于对PANI插层FeOCl的晶体结构和PANI在层间分布的表征,并结合前期的一维电子密度研究,Wu等提出插层过程中苯胺的孤对电子会转移至FeOCl的导带,使苯胺在层间聚合,氨基与FeOCl端位的Cl原子配合促进PANI在层间配位的稳定化(图4)。PANI插层FeOCl在空气中暴露会逐渐解体并发生相变,生成PANI和β-FeOOH的混合物。苯胺在FeOCl层间的聚合、客体-主体配位和构象转化等作用会对FeOCl的电、磁性质造成明显影响。Prassides等[42]利用中子飞行时间技术测量了PANI和PANI插层FeOCl的声子谱,表明层间PANI会形成p型半导体,其导电行为受FeOCl主体的影响。Hwang等[43]利用高分辨中子衍射研究了PANI插层FeOCl的磁结构变化,他们发现PANI插层阻断了相邻FeOCl层结构之间的磁关联,使得FeOCl主体中的Fe自旋呈现出耦合的准二维磁性体系特性,但是PANI的插层对Fe自旋的有序化温度没有明显的影响。
除了苯胺外,研究人员对吡啶和吡咯等杂环芳香胺的插层也进行了许多研究。它们与FeOCl的反应行为和苯胺相似,反应过程的溶剂环境会对插层过程产生较大影响,一般在无溶剂(胺类蒸气)或有机溶剂(醇类、乙腈)插层产物较为稳定,而水溶液环境会极大地阻碍插层反应的进行,而且会导致插层FeOCl结构的解体(生成γ-FeOOH)[44]。这些杂环胺类与FeOCl的摩尔比一般低于1/3,但是戴耀东等[45]曾利用水热法制备了8-羟基喹啉(HQ)插层的FeOCl,其中HQ与FeOCl的摩尔比高达0.78/1。另外,杂环胺类插层可能会对FeOCl的稳定性有较大的影响,Yamada等[46]研究了2-乙烯基吡啶等客体物种在FeOCl中的插层热稳定性,结果显示这些插层化合物在低于423 K就开始分解,较纯相FeOCl的热分解温度(773 K)大大降低,分解产物为Fe2O3、Fe3O4和FeCl2。Kanamaru等[47]曾利用EPR对吡啶插层的FeOCl进行了表征,结构显示在g=2.003处出现了明显的特征峰,对应着吡啶中N孤对电子向FeOCl导带的转移,在其他研究中利用IR、穆谱等也观察到了主-客体之间电荷转移和客体物种聚合的现象,这表明杂环胺类化合物的插层机理与苯胺类似[48]。
作为最为经典的一类FeOCl插层客体,胺类化合物插层的FeOCl结构较为稳定,且插层对FeOCl的基本物理化学性质和层结构都有明显的改变,主要引起了FeOCl导电性的增加、磁各向异性的产生和骨架Fe(Ⅲ)物种的还原,另外胺类物种在层间的聚合还会影响插层化合物的导电性,这些性质的变化使得胺类插层的FeOCl体系在FeOCl插层化合物的研究中具有相当重要的地位,在各类功能材料和器件的开发设计中具有独特的优势。
3.3.2 茂金属插层化合物
茂金属是一种由过渡金属与环戊二烯配位所形成的有机金属化合物,在现有的研究中发现二茂铁、二茂镍、二茂钌、二茂钴和二茂铬等多种茂金属都可以插入FeOCl层间,其中二茂铁的研究最为广泛。茂金属插层会显著提升FeOCl的导电性,如二茂铁插层FeOCl较原始FeOCl导电性大约提升4个数量级[49]。茂金属插层也会造成FeOCl晶体结构的变化,Halbert等[50]发现二茂铁和二茂钴插层的FeOCl层间距拓展了大约5 Å,同时其层结构发生位移,变为体心正交晶系,但是没有证据表明茂金属在层间呈有序化排布。穆谱表征结果表明不同茂金属的插层对FeOCl中Fe位点化学环境的影响基本一致,在FeOCl主体中观察到了Fe(Ⅱ)的生成,部分Fe位点仅与Fe(Ⅲ)配位,而另外的Fe位点中至少观察到与一个Fe(Ⅱ)配位 [51]。二茂铁插层FeOCl的奈尔温度为75 K,在245 K以上,Fe(Ⅱ)和Fe(Ⅲ)呈现出离域特性,发生明显的电子跃迁;而在100~245 K的范围内Fe位点的环境介于高温下离域和低温下局域特征的中间态[51]。Schäfer-Stahl等[49]发现在室温(295 K)下二茂铁插层FeOCl的穆谱仅显示出Fe(Ⅲ)信号,这可能表明FeOCl的端位Cl原子层可能会对电子的跃迁产生屏蔽作用,从而阻碍室温下电子向Fe(Ⅲ)位点的转移。Siu等[52]利用穆谱、X射线光电子能谱(XPS)和电子顺磁共振(EPR)对二茂铁插层FeOCl的表征显示二茂铁在插入过程中被完全氧化,导致主体Fe原子呈现出明显的低自旋-自旋弛豫。
茂金属物种的插层对FeOCl结构和物性的改变与胺类物种相近,但是由于茂金属的金属中心与FeOCl中Fe物种之间可能形成电子相互作用或磁相互作用(电子跃迁、磁耦合等),这使得对于其性质变化的理解更为复杂。另外,层间的茂金属中心也可能成为催化反应的活性位点,因此对于层间环境的探测和设计在茂金属插层的FeOCl体系中需要格外关注。
3.3.3 碱金属阳离子插层化合物
碱金属阳离子的插层一般需要碱金属阳离子的还原性盐(醇盐、碘盐等)与FeOCl反应实现,在插层过程中还原剂诱导FeOCl中Fe(Ⅲ)部分还原,碱金属阳离子作为电荷补偿插入层间。其插层反应方程式如下(以碘化物为例):
Maguire和Banewicz等[53,54]曾以碱金属阳离子的甲醇盐和碘盐与FeOCl作用制备了碱金属阳离子插层的FeOCl,碱金属阳离子与FeOCl的摩尔比在0.16~0.13∶1的范围内。Banewicz等还发现离子直径较小的Na+、K+插层化合物层间距的拓展甚至大于离子直径较大的Rb+、Cs+插层化合物,这可能是由于直径较小的碱金属阳离子会与水或甲醇形成溶剂壳所致。Sagua等[37]研究了正丁基锂与FeOCl的插层,他们发现插层后的FeOCl虽然仍具有半导体性质,但是导电性提升了2~3个数量级。Li的插层对FeOCl晶体结构产生了巨大的改变,诱导生成了长周期的超结构,但是整个插层化合物的结构并不均匀,这可能与正丁基锂在层间的插层、扩散行为有关。
碱金属物种在尺寸和结构上与胺类物种和茂金属存在较大的差异,插层化合物的稳定性与碱金属阳离子的尺寸和溶剂化程度直接相关。由于碱金属阳离子层间配位形式单一,较容易获得明确的晶体学结构,使得对于其层间环境的探测和认识都较为方便,这有助于FeOCl层间环境的开发以实现相应的功能性应用。
3.3.4 烷氧基插层化合物
烷氧基化合物的插层曾由Choy等[39]系统研究,他们利用甲醇盐和乙醇盐与FeOCl反应实现了这一过程。XAS表征显示插层极大地提升了FeOCl中Fe六配位结构的对称性,接近纤铁矿(γ-FeOOH)的配位结构。因此,他们认为烷氧基化合物并不是简单插入FeOCl的层间,而是发生了局部置换反应,部分的烷氧基取代了Cl原子与Fe配位,这也说明了客体物种的插层可能导致FeOCl端位Fe—Cl键的弱化。Kikkawa等[55]将甲醇与FeOCl在110 ℃下浸泡6 d,也制备了甲氧基插层取代的化合物,其仍基本保持层状结构,但是层间距拓展至9.97 Å。IR表征中C—O振动的出现和O—H振动的消失证明了局部取代的发生。在同样的制备方法下将甲醇换为乙二醇也可以发生这种插层取代反应,原始插层产物的层间距为14.5 Å,但用丙酮洗涤后层间距收缩至10.98 Å,说明了这种插层取代结构并不稳定 [56]。
总的来说,烷氧基化合物插层所引起的FeOCl局部置换反应与其他插层客体相比存在较大的差异,这可能与烷氧基物种自身的性质有关(如烷氧基以有机负离子形式存在,具有一定的碱性),对此仍需要进一步的认识。
3.3.5 其他杂环化合物
Kauzlarich等[57]研究了四硫富烯(TTF)、四甲基四硫富烯(TMTTF)、四硫萘(TTN)、四硫并四苯(TTT)等多种有机硫化合物与FeOCl的插层反应,客体与FeOCl的摩尔比随客体物种的尺寸在1/8.5(TTF)到1/23(TTT)范围内变化。XRD显示除了TMTTF与FeOCl层结构平行排布以外,其余有机硫化物均与FeOCl层垂直。有机硫插层后的FeOCl仍具有半导体特性,但电导率是原始FeOCl的103~105倍。IR和XAES表征表明这些客体物种在层间以自由基阳离子的形式存在,与FeOCl的层结构有一定的倾角。Bringley等[58,59]研究了多种多环芳烃(二萘嵌苯、并四苯、苝等)在FeOCl中的插层,这些多环芳烃插入后都与FeOCl层结构平行,IR表征显示其在层间以有机阳离子的形式存在,据此推测每个多环芳烃分子向FeOCl转移1单位的电子,这些插层化合物的电导率较原始FeOCl也有了明显增加,大约提升了105倍。
3.3.6 FeOCl插层体系总结
FeOCl的插层机理和典型的FeOCl插层体系概括于图5。FeOCl主体与客体物种之间氧化还原反应的发生是FeOCl插层过程的典型特征,也是插层能够进行的驱动力,这主要是由于FeOCl中Fe(Ⅲ)/Fe(Ⅱ)与客体物种中氧化还原电对的电势所决定的。伴随着氧化还原反应的发生,客体物种嵌入FeOCl层间,形成插层化合物。根据客体物种尺寸和物理化学性质的差异,FeOCl的晶体学结构和本征的电磁学性质都会发生明显的变化,这些已经在上文中进行了具体的介绍。插层引起的FeOCl性质变化为其在应用领域的开发提供了新的机遇。尽管FeOCl的插层体系研究已经有了大量的文献报道,但是这些研究主要集中在基础的超分子化学领域,缺乏应用导向的结构-性质认识。另外,这些研究主要集中在20世纪中后期,缺乏基于原位表征手段的FeOCl插层结构的动态演化信息,这都需要在今后的研究中予以重视和解决。
4 FeOCl及其插层化合物的应用
4.1 Fenton氧化催化剂
4.1.1 纯相FeOCl体系
Fenton氧化反应即利用Fe基材料活化H2O2生成羟基自由基(HO·)等高氧化性的活性氧物种(ROS),实现污染物的氧化降解,Fenton氧化作为一种主要的高级氧化工艺(Advanced Oxidation Process, AOP)在工业废水、土壤修复等领域具有重要的意义。2013年,本课题组第一次发现FeOCl具有优异的Fenton活性,在典型Fenton反应条件下(pH=4,室温)其活化H2O2生成HO·的能力比传统铁(氢)氧化物高出1~3个数量级,甚至高于均相Fenton体系(FeClO4-H2O2)(表1),对苯、硝基苯等难降解有机污染物都有很好的降解效果[9]。基于高分辨TEM、选区电子衍射等表征,我们认为是FeOCl表面独特的(O-Fe-Cl)或(O-Fe-O)线式构型具有较高的配位不饱和Fe位点分布,能够有效促进H2O2的单电子转移活化。在我们的研究之后,Sun等[60]基于部分热解法对FeOCl的制备进行了优化,制备了FeOCl纳米片材料。他们发现FeOCl纳米片在中性条件下也能够保持很好的Fenton活性,并且在多次反应后通过简单的盐酸处理就可以实现其活性的恢复。他们基于EXAFS等表征提出FeOCl纳米片的表面Fe构型可以有效抑制反应惰性的高价Fe中间体生成,并促进Fe(Ⅲ)和Fe(Ⅱ)的快速循环转化(图6)。
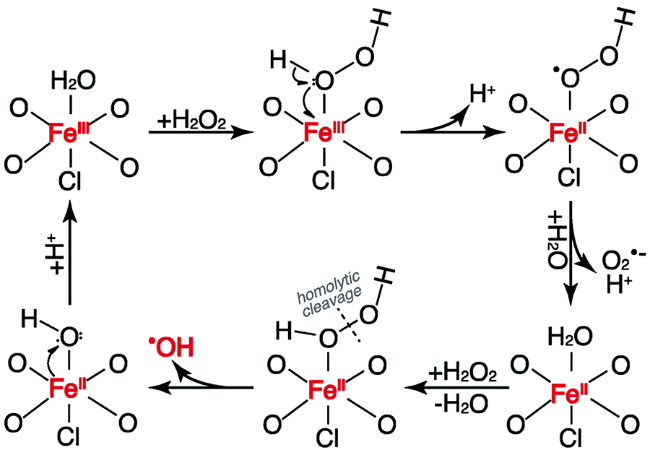
最近,Wang等[61]利用DFT计算详细研究了H2O2在FeOCl表面的分解和转化机理。他们的研究基于FeOCl的(100)晶面,并通过调变表面H原子覆盖度模拟酸性环境。计算结果显示在无H原子覆盖的(100)晶面H2O2很难活化生成HO·,而在H原子完全覆盖的(100)晶面H2O2能够在[Fe(Ⅱ)-O-Fe(Ⅱ)] 结构上活化生成HO·,但是无法从表面解吸。只有在部分H原子覆盖的情况下,首先通过FeOCl质子化和H2O2脱氢的协同作用诱导(100)表面[Fe(Ⅱ)-O-Fe(Ⅲ)]结构生成,这一结构中H2O2既容易在Fe(Ⅲ)位点上活化,而生成的HO·,又容易脱附,这一反应路径可能反映了H2O2在FeOCl表面的真实活化情况。
除了传统的Fenton体系外,外加能量(光辐射、超声)强化也是提升Fenton体系活性的重要途径。ElShafei等[62]曾在紫外光(UV)、超声(US)条件下,以邻硝基苯酚(2-NP)为模拟污染物对FeOCl等多种金属的氧基氯化物的Fenton活性进行了考察。在这一系列催化材料中FeOCl的活性最高,而强化条件对反应活性的影响依次为UV+US>UV>US。在后来的研究中他们还以硝基苯(NB)为模拟污染物对这一体系的反应条件进行了优化,发现可以在较低的H2O2∶污染物投药比(30.86∶1,摩尔比)下实现污染物的高效降解,提升了这一体系的工业应用价值[63]。Li等[64]也对纯相FeOCl的光催化Fenton活性进行了研究。在可见光照射下,这一体系可以在很宽的pH值范围(3~9)对罗丹明B(RhB)进行降解。对HO·的定量分析显示光Fenton体系比传统Fenton体系HO·的生成速率提高了30%。但是总的来说,纯相FeOCl是一种窄带隙(~1.9 eV)半导体,对电子-空穴对的分离效率较低,这限制了其光Fenton体系的进一步开发,研究人员一般会利用其他光催化材料对FeOCl进行改性,以提升其光Fenton性能,这些研究我们将在后面的部分进行介绍。
4.1.2 FeOCl剥离改性体系
将层状FeOCl剥离为少层或单层结构,能够有效提升暴露的Fe活性位点数量,同时Fe位点的反应活性也可能发生变化,这能够提升材料的Fenton活性。Zhang等[17]在乙腈溶剂中通过超声得到了少层FeOCl,展现了比块体FeOCl更为优异的Fenton活性,他们通过DFT计算发现少层FeOCl表面Fe位点拥有比块体FeOCl更低的价态,这可能使少层FeOCl能够更为有效地活化H2O2。此后,Zhang等[65]还基于部分热解法开发了一种“加热-快速退火”的制备方法,直接通过对FeCl3·6H2O的加热剥离制备少层FeOCl纳米片,制备得到的FeOCl纳米片呈FeOCl/氢氧化铁复合结构,和块体FeOCl相比具有较高的比表面积(34.65 m2·g-1)。这种FeOCl/氢氧化铁可以在中性、可见光辅助的条件下实现染料污染物的有效降解,机理研究认为其高Fenton活性的原因是FeOCl/氢氧化铁结构中FeOCl表面的部分Cl被OH取代促进了HO·的生成和脱附。
4.1.3 FeOCl复合材料改性体系
针对纯相FeOCl体系较差的光催化能力,引入高活性光催化材料构筑复合体系,提升FeOCl电子-空穴对分离效率,是促进其光Fenton活性提升的重要手段。Zhang等从杂原子掺杂和异质结构建出发开发了多种FeOCl光催化体系。在杂原子掺杂研究中,他们设计了稀土元素(La和Y)掺杂、Sn4+掺杂和Ce3+掺杂3种体系[66⇓~68],他们以杂原子的氯盐为前驱体通过部分热解法成功将杂原子引入FeOCl的晶格取代位。在光Fenton条件下,以苯酚和布洛芬为模拟污染物,这3个掺杂材料较原始FeOCl都展现出了更加优异的降解活性。DFT计算表明杂原子的掺杂有效地改变了Fe—O、Fe—Cl键的键长,甚至在Sn4+体系中成功诱导了Fe—Cl键的断裂,这使得FeOCl表面Fe位点的化学环境发生改变,电荷量降低,促进了Fe(Ⅲ)和Fe(Ⅱ)直接的转化。
异质结结构是半导体材料层与层结构叠加所形成的一种特殊界面结构,通过形成异质结结构也可以有效提升FeOCl的电子-空穴对的分离效率,进而提升其光催化活性。Zhang等开发了氧化石墨烯(GO)、还原氧化石墨烯(rGO)和多金属氧酸盐(POM)三种异质结体系[69⇓~71],Xie等[72]开发了g-C3N4异质结体系。这些FeOCl异质结体系在以罗丹明B(RhB)和苯酚为污染物的活性考评中也展现出了优异的催化活性,而且由于异质结结构保护了FeOCl的层结构,使得Fe的浸出情况大大降低。基于光致发光(PL)光谱、电化学阻抗谱(EIS)的表征表明异质结体系拥有比FeOCl体系更快的界面电荷转移和更高效的光生载流子分离能力,DFT表征也显示了异质结界面处Fe原子的电荷比原始FeOCl体系更低,这表明界面处Fe原子与H2O2具有更高效的电荷转移速率,是其Fenton活性提升的原因。
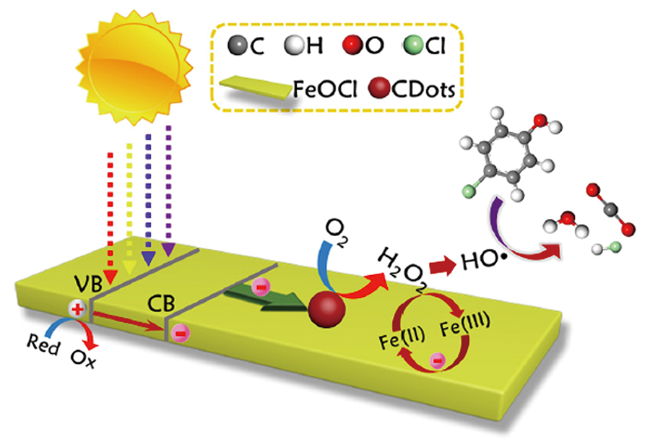
4.1.4 FeOCl插层化合物体系
插层改性可能会对FeOCl几何和电子结构产生明显影响,进而影响其催化性能,基于这种认识,本课题组最近开发了聚苯胺(PANI)插层的FeOCl体系[74]。我们首先在乙腈溶剂下制备了不同插层时间的FeOCl-PANI材料,渗透凝胶色谱(GPC)表征显示随着插层时间的增加,层间PANI聚合度(分子量)逐渐增加。我们将不同插层时间FeOCl-PANI在酸性(pH=4)和中性条件下用于2-甲氧基苯酚(2-MeOP)等污染物的Fenton降解,发现其反应活性均高于原始FeOCl体系。更值得注意的是,在酸性和中性条件下,FeOCl-PANI的插层时间与其反应活性呈现出相反的趋势,即酸性条件时插层时间最短的(4 h)材料拥有最高的反应活性,随着插层时间增加反应活性逐渐降低;而中性条件时插层时间最短的材料反而反应活性最低,随着插层时间增加反应活性逐渐增加,插层时间最长(12 d)的材料活性最高。对此,我们基于EXAFS、穆谱、DMPO捕捉EPR以及吸附实验等方法对FeOCl-PANI体系这一反应特性进行了揭示,在酸性条件下Fenton反应的本征反应性很高,反应体系主要由扩散控制,而污染物会与层间PANI存在氢键相互作用,抑制污染物向反应位点的扩散,插层时间越长的材料PANI聚合度越高,对污染物扩散越不利,因此活性越低;而在中性条件下Fenton反应的本征反应性较低,反应主要由本征反应性控制,聚合度越高的材料PANI与层间Cl原子配位形成的开放Fe位点数量越多,因此活性越高(图8)。这一体系是针对FeOCl层间区域的结构和Fenton反应性探究的第一个案例,可能对FeOCl层间空间的功能性开发利用具有一定的启发。

4.1.5 FeOCl工程化体系
FeOCl在前述的研究中展现出了优异且富于调变的Fenton活性,有许多研究者根据实际的工业应用需求对其进行了一定的改性或与特定工艺耦合,也取得了很好的效果。对此最早的研究是本课题组对FeOCl进行的负载化改性[75,76],为了提升FeOCl材料在实际工业污水处理条件下的实用性,我们开发了浸渍焙烧的方法以实现FeOCl的负载化,浸渍焙烧法是基于部分热解法发展的一种FeOCl的制备方法,主要应用于负载型FeOCl材料的制备。其具体制备流程是将载体组分直接与无水氯化铁(FeCl3)或六水氯化铁(FeCl3·6H2O)的水溶液、醇溶液或熔融(80~100 ℃)液浸渍,再高温加热生成FeOCl物相。在这一过程中FeOCl的相转化过程与部分热解法相同,但由于与载体的相互作用,可以有效地提升FeOCl的稳定性,避免其结构解体或活性组分浸出。
利用上述方法,我们成功将FeOCl负载在介孔二氧化硅(SBA-15)表面,Fe的负载量为10 wt%。制备得到的FeOCl/SBA-15在保持循环稳定性的同时仍具有优异的Fenton活性,对H2O2在这一体系中的分解建立了基元反应模型(式6):
式中的动力学常数可以通过实验数据拟合,计算得到分解H2O2的西勒(Thiele)模数在10-5~10-6 cm2/s范围内,反应属于表面控制过程。进一步地,我们针对这一体系以双酚A(BPA)为模拟污染物建立了半经验的动力学方程,涵盖了各个反应参数对BPA降解的影响,并利用响应面法对该体系的反应参数进行了优化。

4.2 其他高级氧化(AOP)催化剂
除了以H2O2为氧化剂的Fenton氧化体系外,FeOCl也能催化其他过氧化物的活化实现有机物的降解,这些AOP体系的研究近年来也逐渐得到了关注。Li等[81,82]发现在可见光催化下FeOCl可以有效活化过碳酸盐(PC)和过硫酸盐(PS),实现RhB的降解,其中PS体系可以在很宽的pH范围(3~10)内都有明显的催化活性。活性氧物种探测发现HO·和超氧阴离子(O2-·)是污染物降解的主要ROS。Xie等[83]研究了FeOCl对过一硫酸盐(PMS)的活化,发现这一体系在近中性的反应条件下可以对丙咪嗪(Imipramine,一种抗抑郁药)等多种新兴污染物和硝基苯等多种难降解有机物都有很好的降解活性。活性氧物种探测显示体系中存在两种ROS,即硫酸根自由基( ·)和HO·,但是 ·在体系中的浓度更高,是污染物降解的主要ROS。另外反应体系中一些杂质离子(如碳酸氢盐、氯离子等)也可能参与PMS活化的链式反应促进ROS生成,考虑到这些离子在真实水体中十分常见,因此FeOCl-PMS体系可能在天然水体环境的修复当中具有明显的应用价值。在此基础上,Li等[84]将光催化引入PMS的活化体系,通过可见光激发光生电子实现Fe(Ⅲ)/Fe(Ⅱ)的快速循环,这将FeOCl/PMS体系的pH应用范围拓展至碱性环境(pH=11),进一步提升了这一体系的应用意义。
4.3 选择氧化催化剂
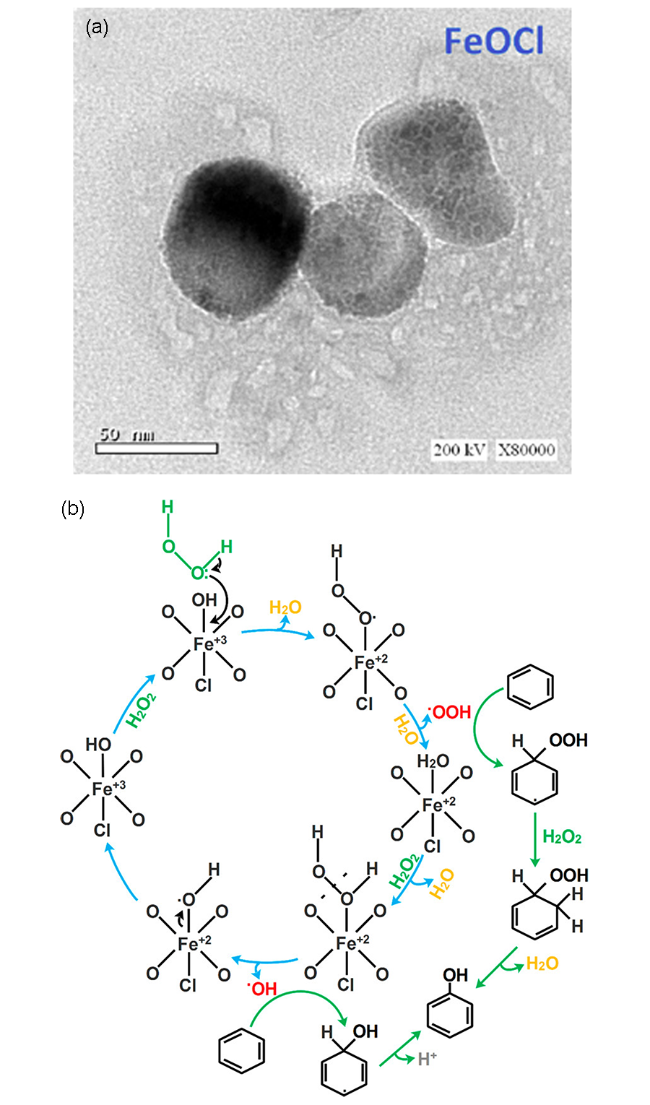
FeOCl在环境催化领域的Fenton氧化等AOP体系中展现出了优异的反应性和发展潜力,这些AOP体系主要依赖生成高的ROS的无差别氧化性实现有机污染物深度氧化降解。除此以外,对氧化过程的强度进行调控,使其能够有选择性地氧化目标反应物生成特定产物,这被称为选择氧化体系,在工业化学品合成等工业催化领域十分重要。Elmetwally等[10]研究了基于FeOCl的苯催化羟基化制苯酚这一选择氧化反应。他们采用化学气相迁移法,基于小尺寸的Fe2O3前驱体制备了FeOCl纳米颗粒,其粒径~50 nm(图10a)。制备的FeOCl纳米颗粒在乙酸为反应溶剂下进行了苯羟基化反应的测试,通过调变反应时间、催化剂/氧化剂投加量和反应温度等工艺参数发现在最优反应条件下苯的转化率~40%,而苯酚的选择性接近100%。他们认为在乙酸溶剂条件下,FeOCl表面丰富的Fe(Ⅱ)位点能够有效激活H2O2生成HO·,HO·与苯发生直接抽氢反应生成苯酚(图10b)。
4.4 有机合成催化剂
除了在AOPs和选择氧化催化体系中应用以外,FeOCl还在一些有机合成催化体系的研究中有见报道。Selvakannan等[85]利用热缩合方法在有序介孔材料MCM-41的表面硅醇基团上接枝了FeOCl纳米物种。表面FeOCl作为Lewis酸性位可以有效催化萘酚、酰胺和醛类的多缩合反应,反应产物烷基萘酚是一种重要的制药中间体。Mohammadi等[86]以BiCl3和FeCl3·6H2O为前驱体通过浸渍-焙烧制备了SiO2负载的BiOCl/FeOCl催化剂(BiOCl/FeOCl/SiO2),可以在无溶剂条件下催化苯甲醛和邻苯二胺合成2-芳基-1H-苯并咪唑,催化剂可以至少循环使用3次活性未见明显下降。
4.5 电极材料
4.5.1 电池电极材料
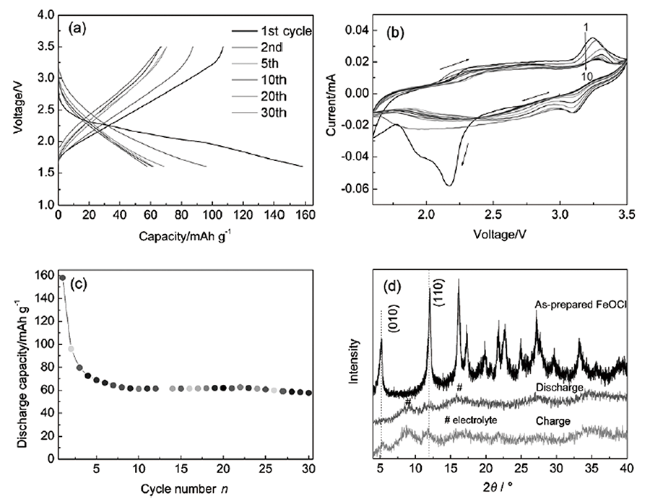
电极材料是FeOCl在催化剂以外的另一主要应用领域。在20世纪70年代,埃克森研究与工程公司(Exxon Research & Engineering Co.)在其公开的一项专利中第一次提出了以FeOCl作为锂离子电池正极材料的可能 [8]。在专利的实施例中,其将FeOCl与炭黑(10 wt%)和聚四氟乙烯(10 wt%)混合,在室温下压制于不锈钢格栅并于300 ℃下焙烧制备成电极格栅。这一材料与金属Li负极一起组成的锂离子电池,开路电压为2.96 V,可以以1~4 mA的放电速率重复充放电100次。1989年,Takehara等[87]也将FeOCl作为锂电池的正极材料进行了研究,这一体系的放电容量是传统二氧化锰材料的3倍左右,但是在充放电测试中发现FeOCl的稳定性较差,在放电过程中Li会插入FeOCl的层间,最终造成FeOCl结构的解体,分解成α-Fe、LiCl和Li2O。此后他们还对苯胺插层FeOCl的充放电过程进行了测试研究[88],发现在充放电过程中苯胺插层FeOCl会变为γ-FeOOH和苯胺衍生物(聚合物或低聚物),其中苯胺衍生物会发生氧化还原,这也表明了材料的不稳定。
以纯相FeOCl构建的CIB虽然获得了成功,但是由于FeOCl导电能力不佳,电荷转移和传质缓慢,造成FeOCl的实际放电容量较低。为了改善FeOCl的导电性,Zhao等将FeOCl与多种碳材料(碳纳米管、炭黑、石墨烯)分别进行机械球磨混合,制备了FeOCl/碳复合材料电极[91],其中石墨烯的加入使得FeOCl电极的电化学性能得到明显改善,其可逆放电容量提升至184 mAh/g。他们还通过DFT计算对FeOCl晶格中不同Cl位点迁移的静电势能进行了计算比较,初步揭示了充放电过程中FeOCl的相变机理。此后,他们又开发了多种FeOCl复合碳基材料体系[92,93],如通过真空浸渍-部分热解的制备方法制备了有序介孔碳CMK-3负载的FeOCl(FeOCl/CMK-3),以及通过部分热解、气相聚合方法制备了聚吡咯(PPy)包覆的FeOCl材料(FeOCl/PPy)。其中FeOCl/CMK-3的可逆放电容量达到了202 mAh/g,而FeOCl/PPy的可逆放电容量达到了187 mAh/g,同时这2个体系都具有很好的稳定性。
除了考虑将FeOCl与其他材料复合,Zhao等[94]还从插层改性的角度考察了FeOCl在CIB中的应用。他们设计了苯胺插层的FeOCl-聚苯胺体系(FeOCl-PANI)电极,PANI插层促进了FeOCl的电荷转移,也能够对FeOCl的层间结构进行稳定,这使得FeOCl的电化学性能明显改善。随着插层时间的增加FeOCl-PANI的可逆放电容量逐渐提升,最大(6 d插层)达到了120 mAh/g(50个循环周期),容量保持率达到了82%,远高于原始FeOCl的42%。
4.5.2 超级电容器电极材料
FeOCl在非对称超级电容器(ASCs)阳极材料的开发中也得到了应用。Wan等[12]通过机械球磨将FeOCl和多孔纤维素衍生碳基气凝胶(CDCA)混合制备了FeOCl/CDCA材料,CDCA的引入使得FeOCl的机械稳定性和电荷存储动力学都得到了明显的增强。FeOCl/CDCA在2 mA·cm-2的电流下可以提供高达1618 mF·cm-2(647 F·g-1)的超高比表面积电容,同时在1 M Na2SO4 电解液、-1到0 V的电压范围内经过10 000次循环后电容损失在10%以内,展现出了极高的稳定性。使用FeOCl/CDCA与MnO2阴极组成的ASC展现出了很高的能量和功率密度(289 μWh·cm -2 、1.8 mW·cm-2)以及优异的倍率特性和循环稳定性。在这一体系中FeOCl的层状堆叠结构具有较大的比表面积、较高的理论电容值和良好的力学稳定性,同时高导电性、多孔的CDCA能够作为电解质体系缩短离子与层状FeOCl之间的离子扩散路径(图12),这些特性决定了FeOCl/CDCA在ASC中的优异电化学性能。
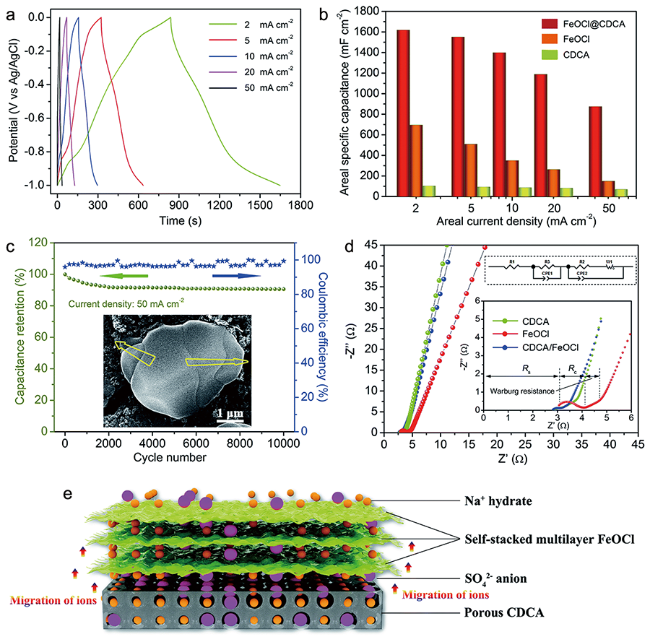
图12 FeOCl/CDCA电极的电化学性能:电流密度为2~50 mA·cm-2下的恒电流充放电(GCD)曲线(a);倍率性能(b)50 mA·cm-2下的Nyquist点图(内图是高频区域放大像)(c);EIS数据拟合的等效电路(d)和FeOCl/CDCA组分在电化学反应中的功能示意图(e)[12]Fig. 12 Electrochemical properties of the FeOCl/CDCA electrode: GCD curves of FeOCl/CDCA at current densities of 2~50 mA·cm-2(a); rate performance(b); Nyquist plots of FeOCl@CDCA at 50 mA·cm-2 and insets show the enlarged image at the high-frequency region(c), the equivalent circuit used for EIS data fitting(d); schematic diagrams of functions of CDCA and FeOCl components on the electrochemical reactions(e)[12] |
4.6 其他应用
除了作为催化剂和电极材料,FeOCl还在其他领域有一些应用研究。Sun等[95]曾对FeOCl通过超声剥离制备了二维FeOCl纳米片,展现出了对水环境中Pb(Ⅱ)的很好吸附效果,吸附容量能达到409.71 mg/g,超过目前绝大多数吸附材料,且吸附平衡时间仅为块体FeOCl的三分之一。HRTEM和DFT计算显示二维FeOCl纳米片主要暴露(110)晶面,Pb(Ⅱ)在该晶面的吸附比FeOCl其他晶面具有更低的吸附能,能形成更为稳定的吸附结构从而有利于Pb(Ⅱ)的吸附。Wang等[96]对FeCl3溶液利用液体脉冲激光烧蚀(LAL)成功制备了FeOCl纳米片,再将Au纳米颗粒均匀修饰在FeOCl纳米片表面。由于Au对HCl的选择吸附,这一Au/FeOCl复合纳米材料可以作为化学电阻在室温下高敏感度和高选择性实现空气中HCl的感测相应,可以应用于工业场所的HCl检测。LAL法的主要原理是利用在短时间内产生的高能量脉冲激光引起前驱体组分的相变,这种方法在微/纳米结构化合物的合成中已经有了十分广泛的应用[97]。在这里,由于脉冲激光的定向性和高能性,使我们可以更为可控地实现FeOCl的相转化,即只要改变FeCl3溶液的浓度就可以有效调节Au/FeOCl材料的晶体大小和组成,这是其较传统的部分热解法或化学气相迁移法最为明显的优势。但是其激光装置需要特定设备且能耗较高、对于反应条件的要求较为苛刻、难以规模化放大,这些缺点限制了LAL法在FeOCl制备中的进一步应用。Schoop等[98]通过正丁基锂插层,并在甲醇溶剂中通过对体系湿度的精确控制实现了大尺寸(0.5~1.0 μm)FeOCl二维纳米片的剥离,AFM显示其厚度仅为~2.4 nm( 图13a,13b),对其磁化率的测量显示FeOCl二维纳米片具有和块体FeOCl一样的反铁磁特性,但是其磁化率与温度的关系遵循居里-韦斯(Curie-Weiss)定律,具有较低的居里温度,且具有磁阻挫现象(图13c,13d)。这种少层FeOCl纳米片作为一种新颖的二维反铁磁体可以应用于自旋电子器件的开发。
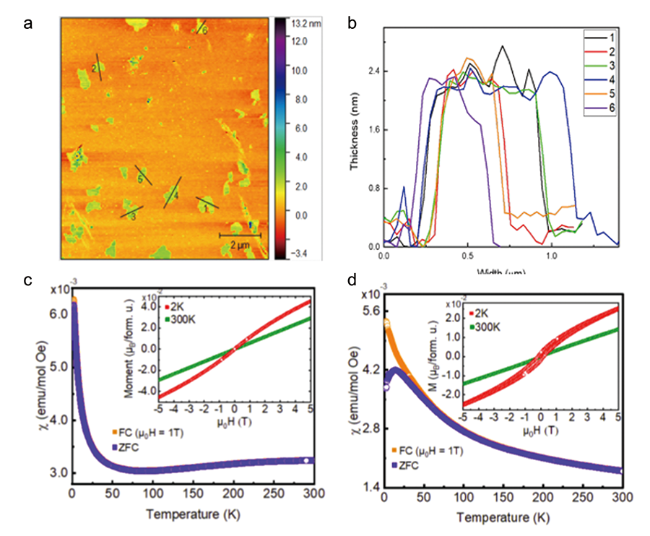
另外,FeOCl也可以作为前驱体或模板剂,诱导其他材料的合成。Li等[99]首先通过化学气相迁移法制备了块体FeOCl,再将其超声剥离为FeOCl纳米片,最后利用溶剂热还原的方法诱导FeOCl相转化得到Fe3O4纳米带(nanobelt)。制备得到的Fe3O4纳米带有着很高的磁饱和值(~53 emu·g-1)和磁各向异性(6.6 × 10 4 erg·cm-3),能在大多数溶剂环境中均匀分散,在生物医药材料中具有广泛的应用前景。利用FeOCl通过相转化也可以制备α-Fe2O3,Frydrych等[100]和Marinović-Cincović等[101]分别用高温焙烧和液相老化成功实现了这一转化过程。Yang等[13]利用化学气相沉积法(CVD)在掺氟氧化锡玻璃衬底上制备了FeOCl纳米片阵列(NSAs),再通过焙烧-退火将其转化为多孔α-Fe2O3 NSAs(图14),这一材料的电势高达1.23 V(vs RHE),是普通α-Fe2O3的3倍,可以用于光催化水解离。Xu等[14]利用FeOCl为模板通过层间限域原位聚合-酸洗成功制备了超薄聚吡咯(PPy)纳米片,其可以作为第二红外区域的光热剂应用于肿瘤消融等医学治疗过程。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
近年来,基于FeOCl的各类应用研究展现出蓬勃的发展趋势,其中催化剂研究是FeOCl发展最为迅猛的一个领域,各种基于H2O2和其他过氧化物活化的AOP体系被广泛开发,一些选择氧化体系也逐渐发展,这体现了研究人员对氧化剂在FeOCl表面活化行为认识的深入和调控手段的进步。另外,FeOCl作为电极材料在CIB和ACS中的应用也日益增多,成为继催化剂之后第二大应用研究方向。但是,我们也必须看到FeOCl日益蓬勃的应用研究与其前期大量的基础研究是分不开的。FeOCl制备的可控性和层结构可调变性是其应用体系开发的基础。对FeOCl制备方法(化学气相迁移法、部分热解法等)和晶体学结构认识的深入使我们能实现FeOCl形貌、尺寸的有效调控,而对FeOCl插层行为的认识和各种插层体系的开发能使我们通过不同的插层过程,实现FeOCl晶体学结构和电子结构的有效改变,因为不同的插层客体具有不同的几何尺寸、电荷量。另外,不同有机(金属)客体物种的插层会在FeOCl层间形成特定配位结构的有机-无机杂化界面[102,103],层间端位Cl原子会与客体物种以不同的结构配位,从而对Fe—Cl键的化学环境和层间的空间分布造成显著影响。基于此,我们可以定向设计表面Fe位点的几何和电子结构、FeOCl层间结构,从而开发更为高效、实用的催化体剂或开发具有优异电化学性能和循环稳定性的电极材料。
然而,在FeOCl体系的研究当中仍然有一些重要的问题需要进一步解决。第一,我们需要开发更为可控的FeOCl制备技术,以实现特定晶面(特定生长方向)FeOCl的制备,这需要我们对制备过程中FeOCl的物相转化过程有更深入的认识;第二,我们需要对FeOCl的几何和电子结构有更为深入的了解,进而实现其表面Fe位点反应性(催化领域)或FeOCl结构氧化还原性能(电极材料领域)的精确调控,可控的插层改性是目前最有希望实现这一目的的手段,但是可能还需要和先进的表征技术或理论计算相配合;第三,由于FeOCl的层状结构和Fe-Cl键的离子键特性,其稳定性比一般铁(氢)氧化物稍差,需要开发高稳定性的FeOCl体系,这需要我们明确FeOCl在原位工况条件下的结构演化行为,并在提升前两点认识的基础上进行解决,如开发负载型FeOCl材料、诱导高稳定晶面的生成、通过插层等手段强化层间范德华作用力等。
总的来说,虽然我们目前已经对FeOCl的物理化学性能有了一定的了解,但是这些认识还比较浅显,特别是如何建立FeOCl结构与其性能的关系仍存在许多争议和不确定性,我们需要进行更加深入的研究。FeOCl是一类新兴的Fe基层状材料,围绕其科学研究还是应用开发都还有许多有趣但艰巨的任务,但我们相信随着对其认识的逐渐深入,特别是对FeOCl插层化学行为和其插层化合物基本性质和应用研究的深入,FeOCl体系将在环境、能源等领域做出重大的贡献。