




1 引言
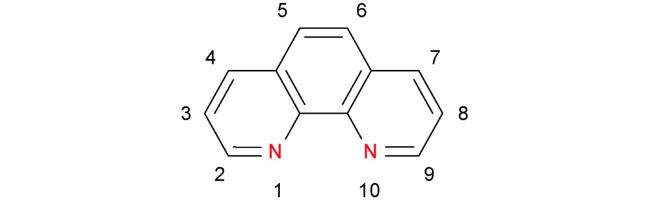
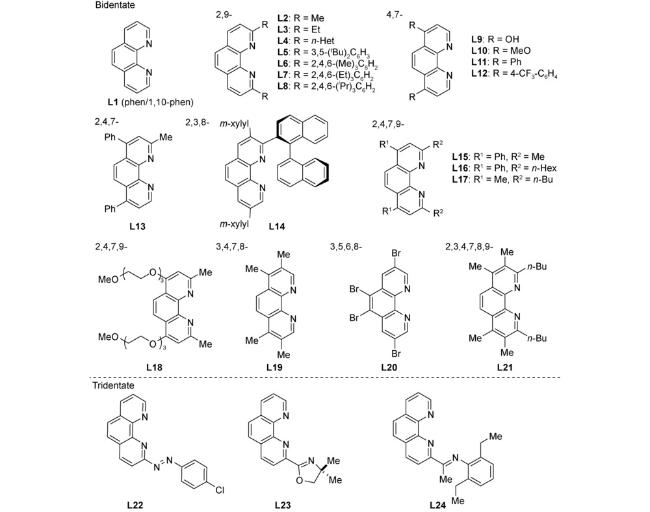
2 邻菲罗啉类配体在铁催化反应中的应用
2.1 偶联反应
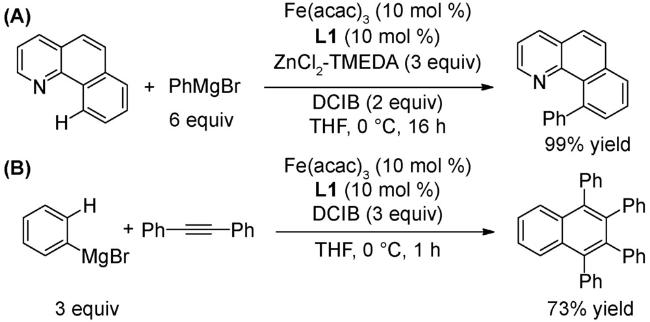
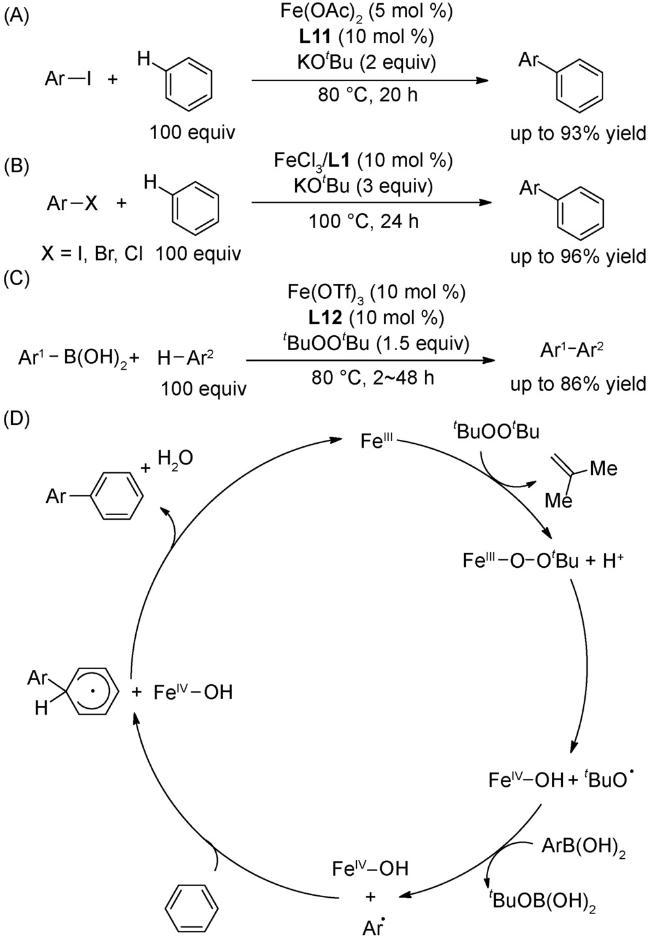
图式2 (A)芳基自由基转移芳基化[34];(B)非活化芳烃的直接芳基化反应[35];(C)芳基硼酸和苯衍生物的氧化偶联反应[36];(D)式(C)可能的机理[36]Scheme 2 (A) Direct arylation through an aryl radical transfer pathway[34];(B) Direct arylation of unactivated arenes[35];(C) Oxidative coupling of arylboronic acids with benzene derivatives[36];(D) Proposed mechanism of(C)[36] |
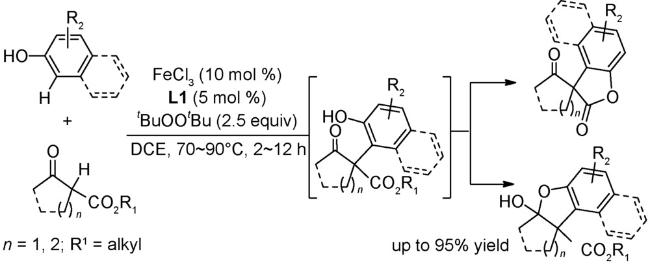

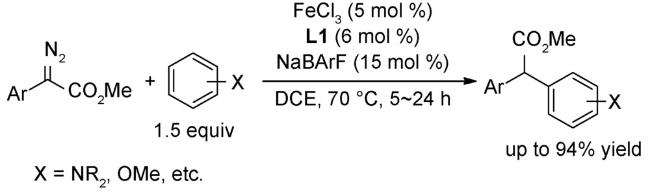
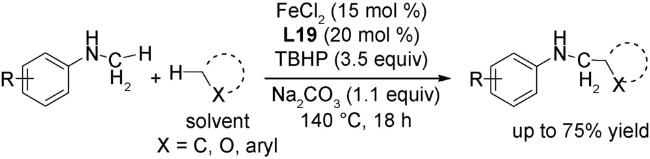
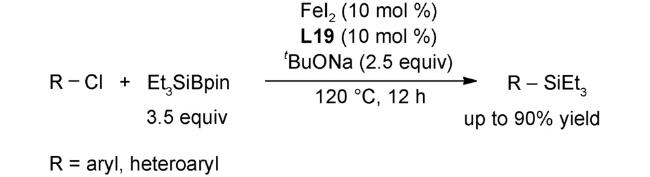
2.2 氧化反应
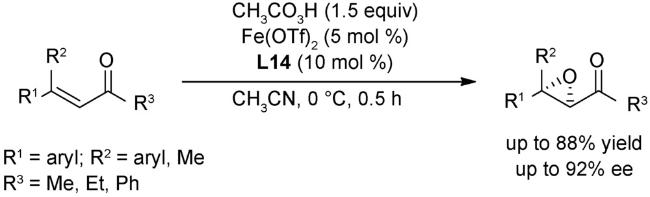
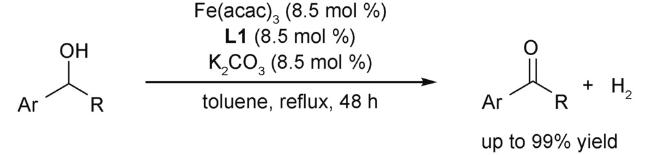
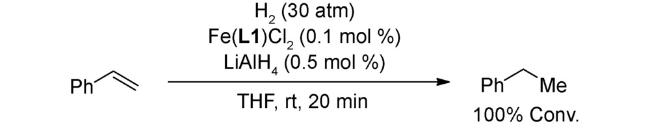
2.3 还原反应
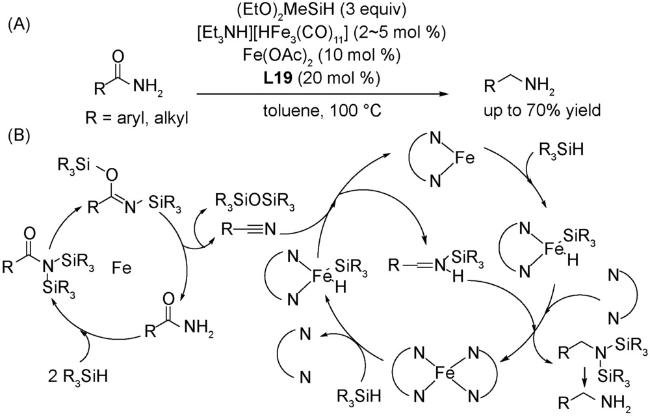
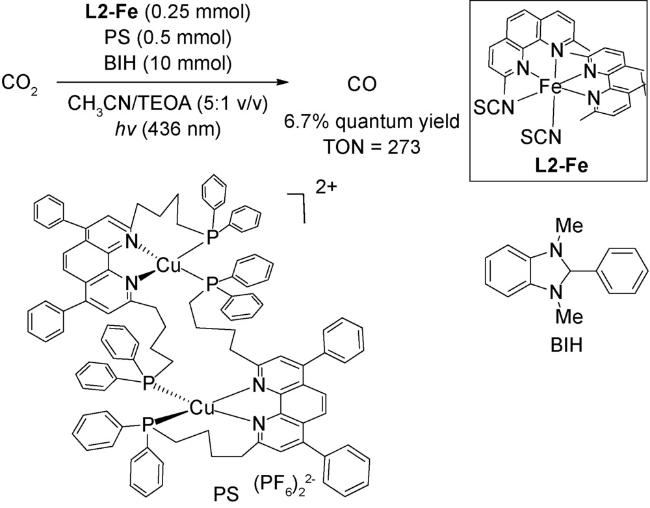
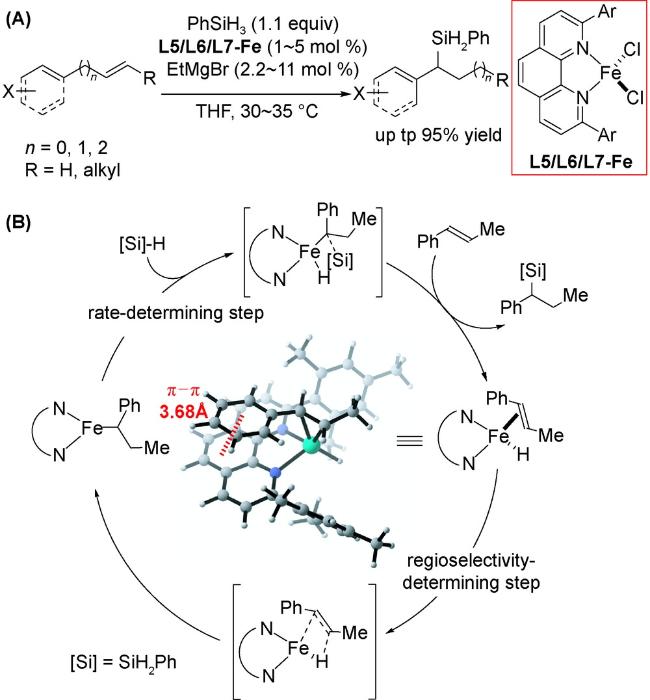
2.4 加成反应
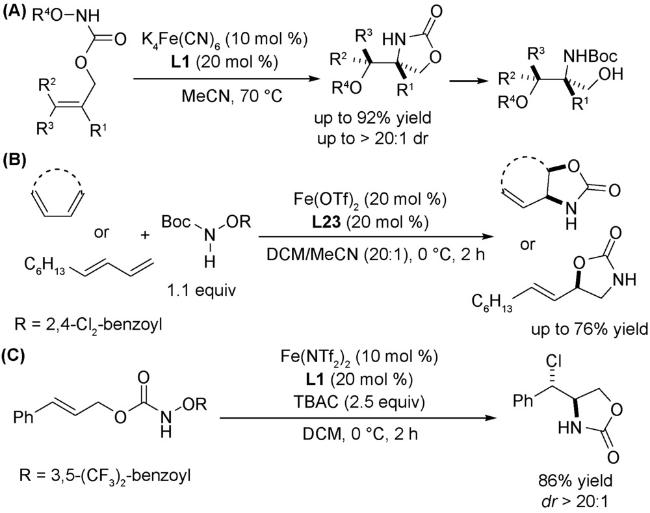
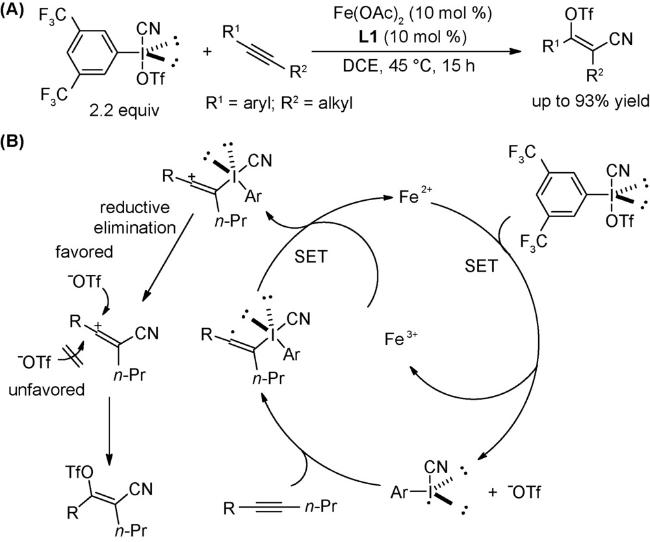
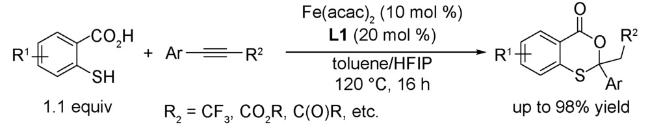
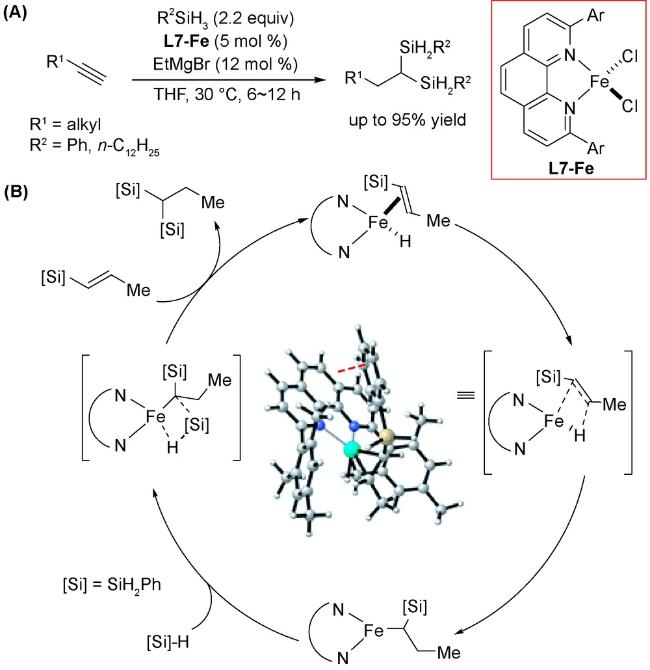
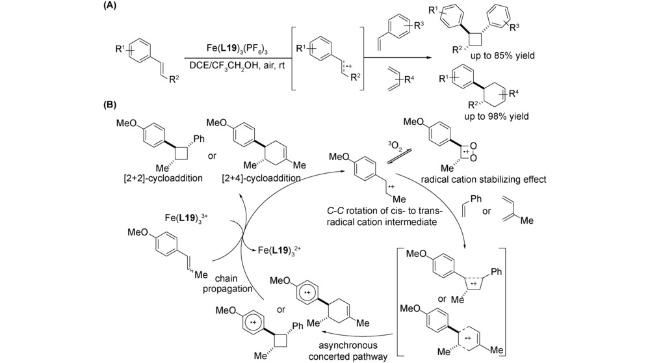
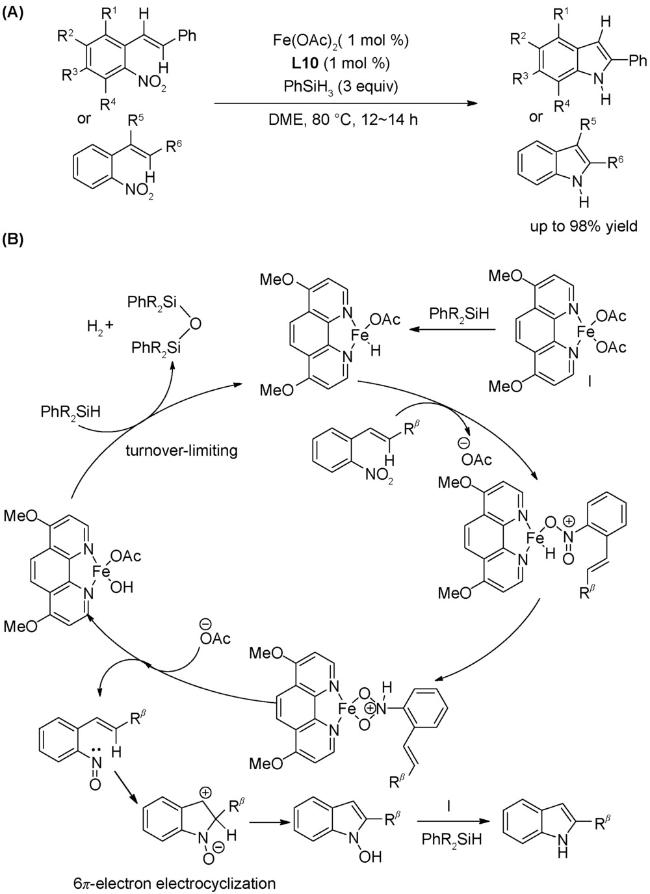
2.5 其他反应
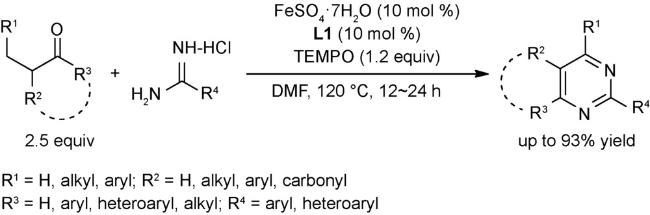
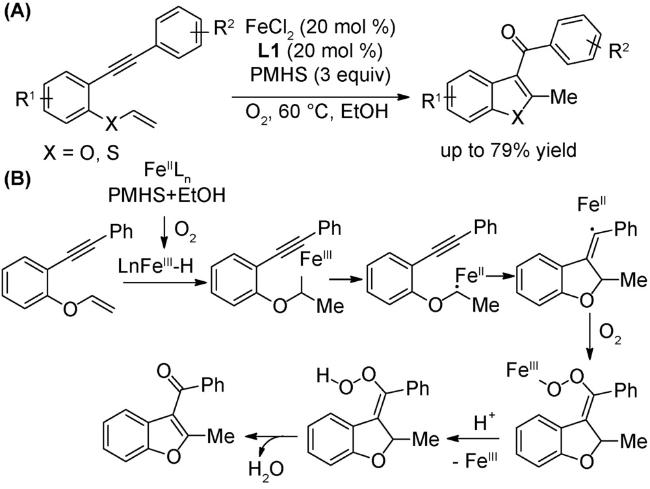
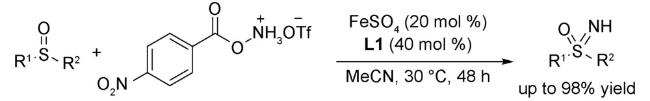
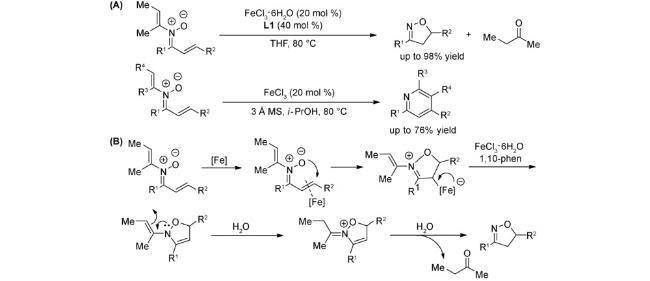
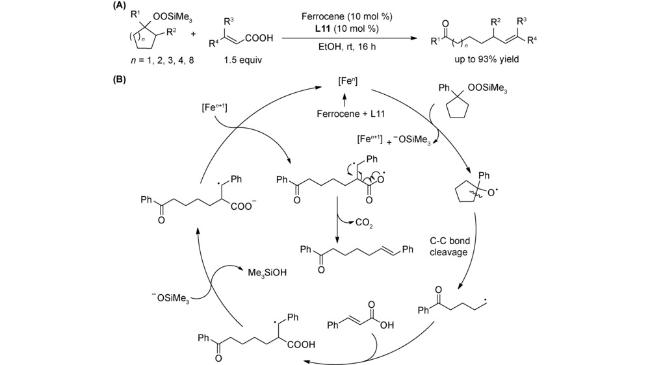
3 邻菲罗啉类配体在钴催化有机反应中的应用
3.1 加成反应
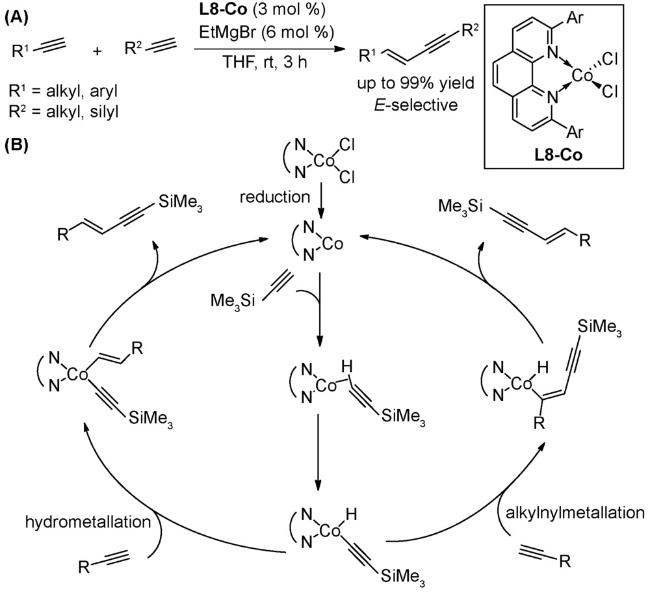
3.2 环加成反应
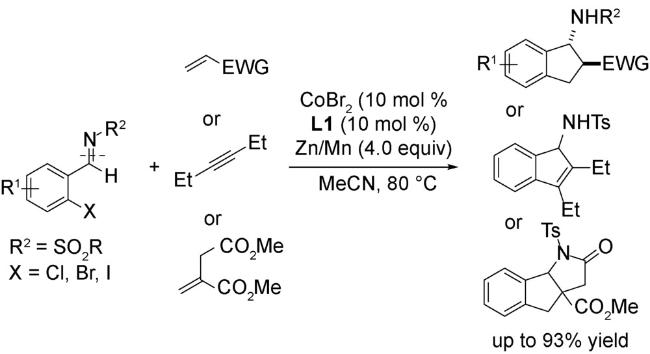
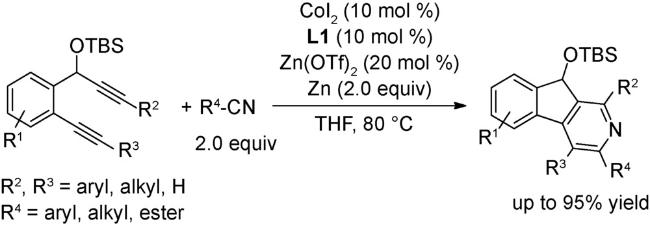
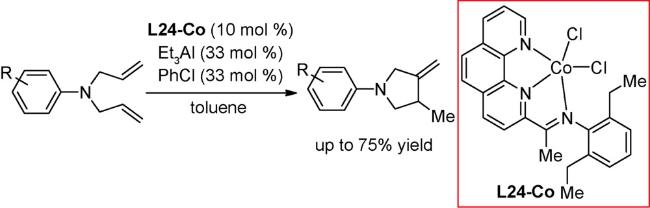
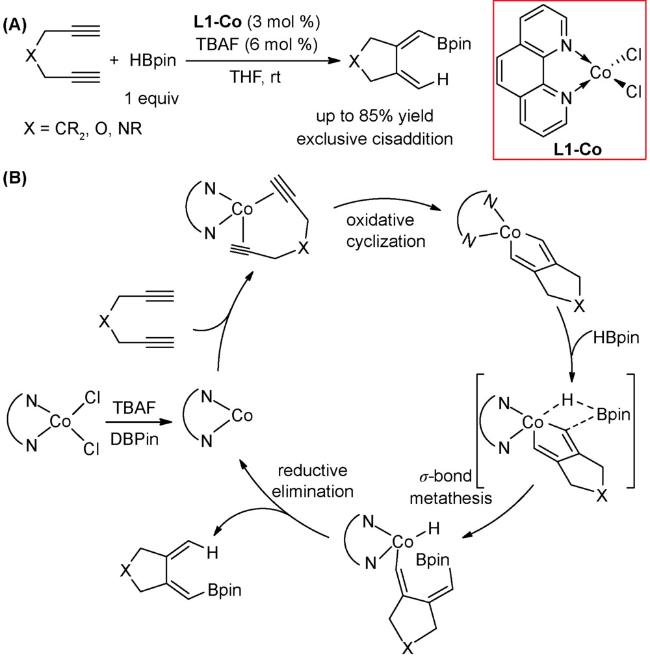
3.3 C—H键官能团化
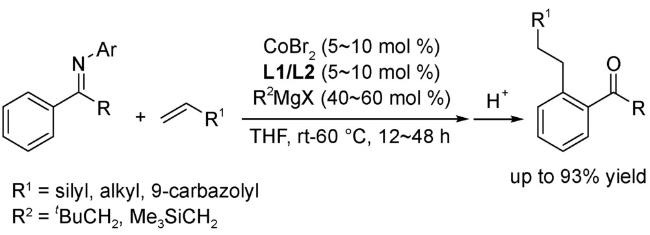
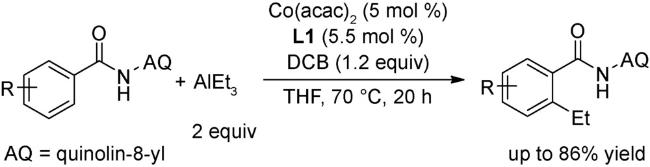
3.4 羧化反应
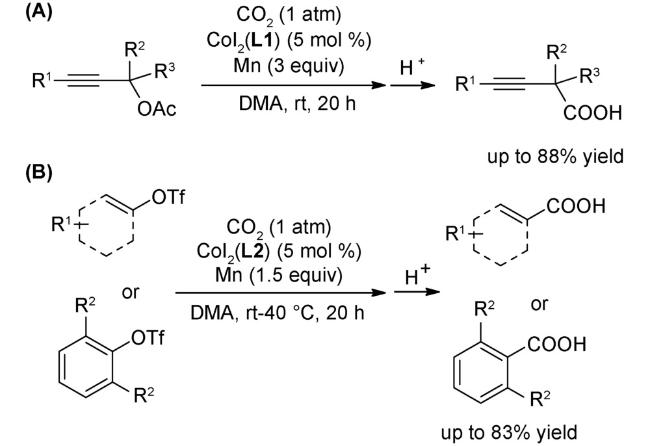
3.5 偶联反应
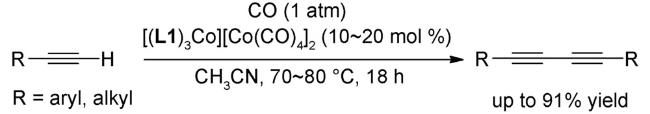
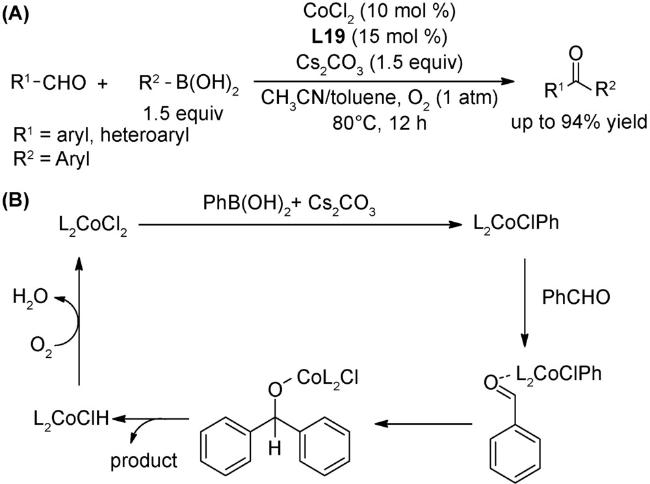
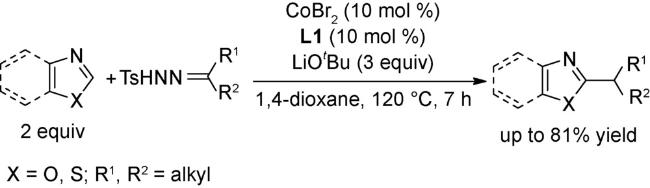
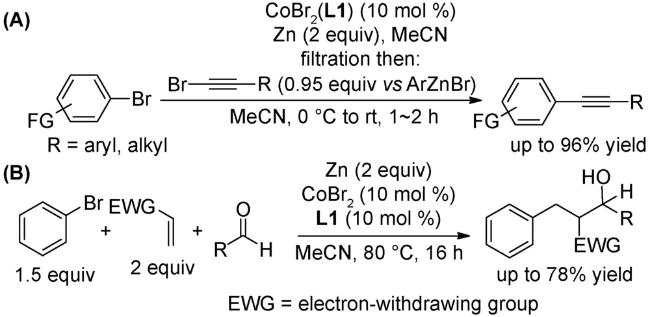
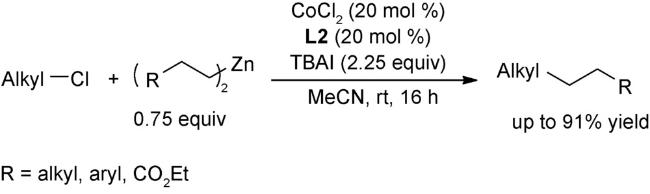
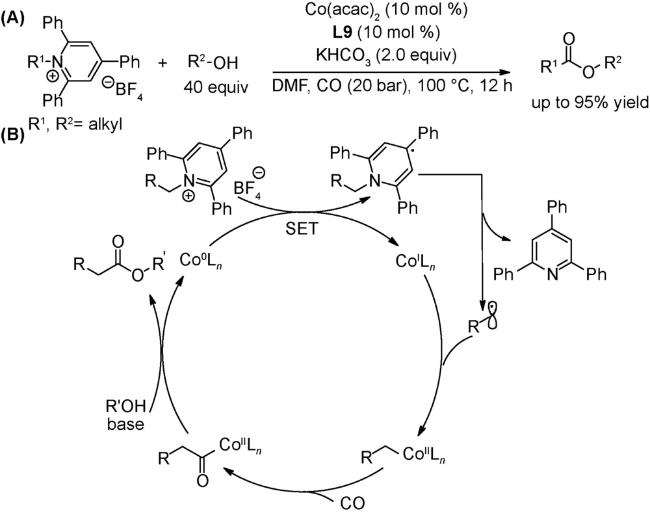
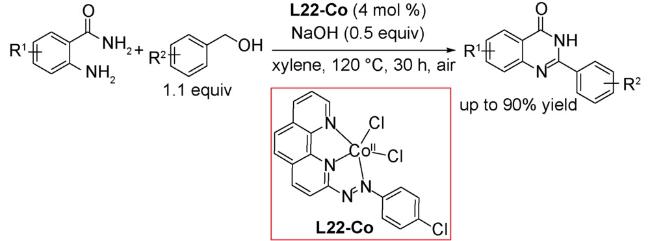
3.6 其他反应
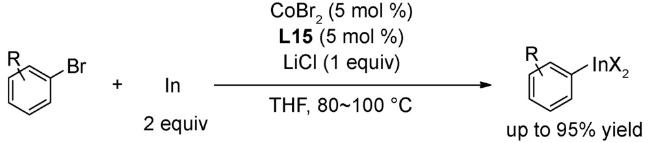
4 邻菲罗啉类配体在镍催化反应中的应用
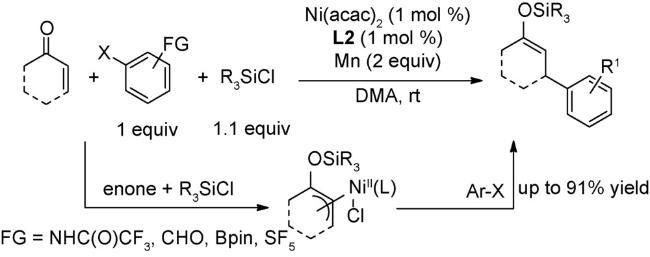
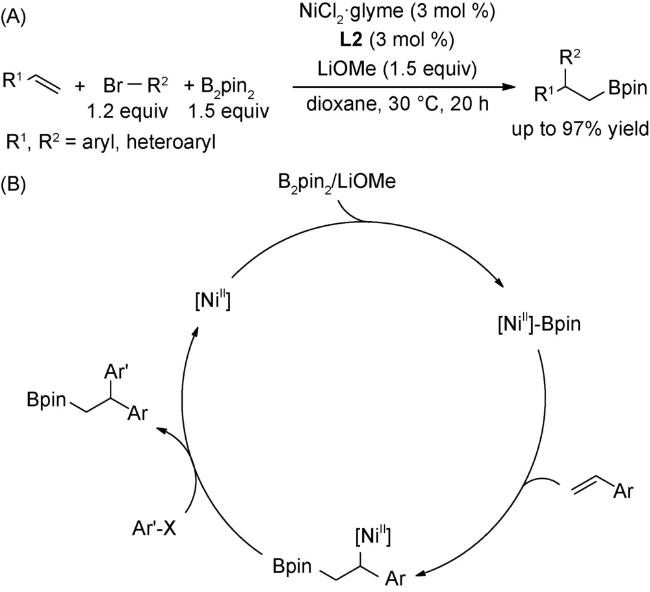
4.1 交叉偶联反应
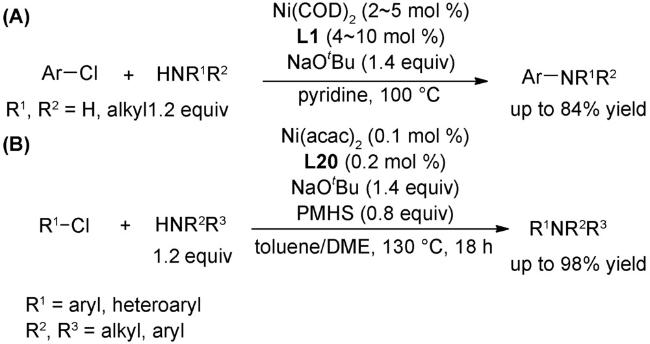
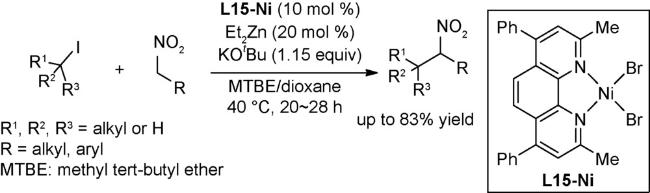
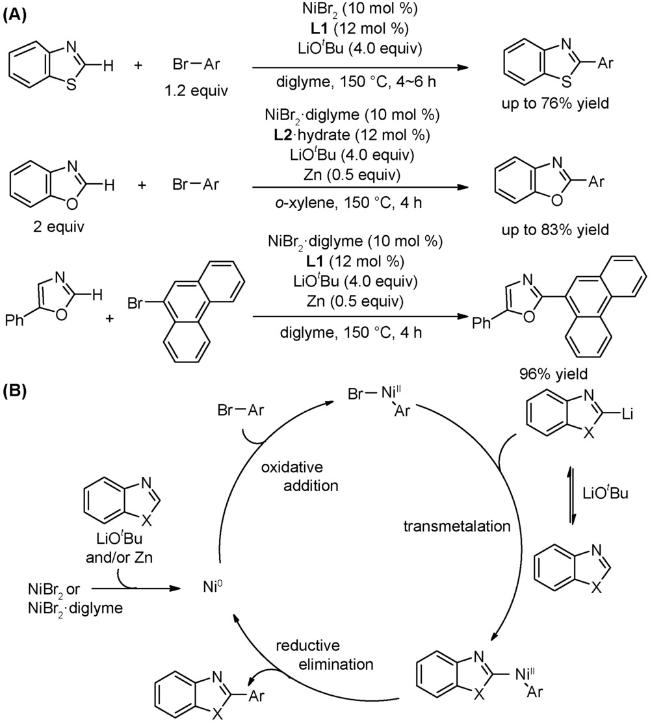
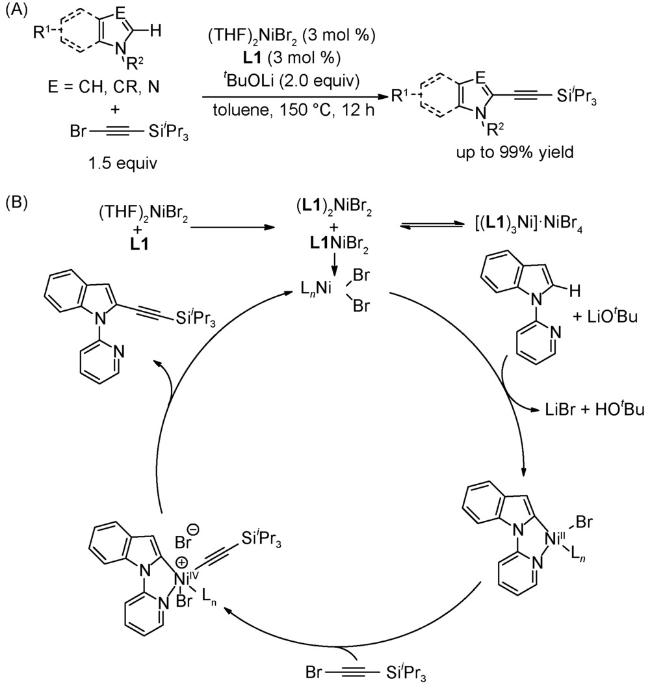
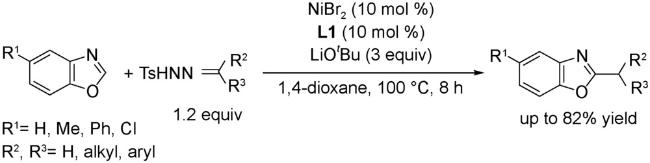
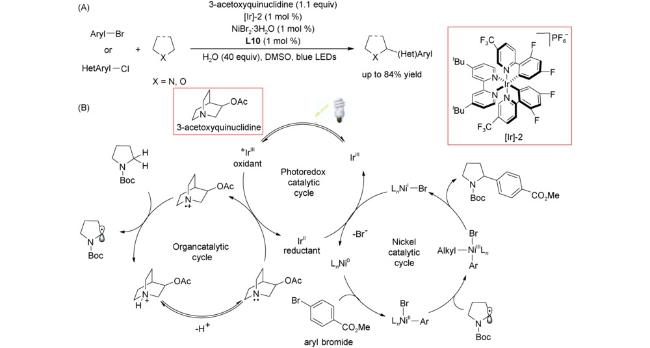


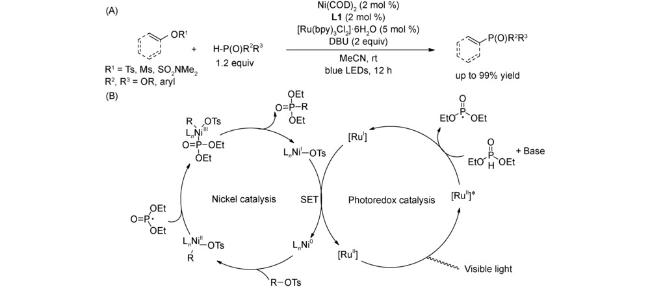
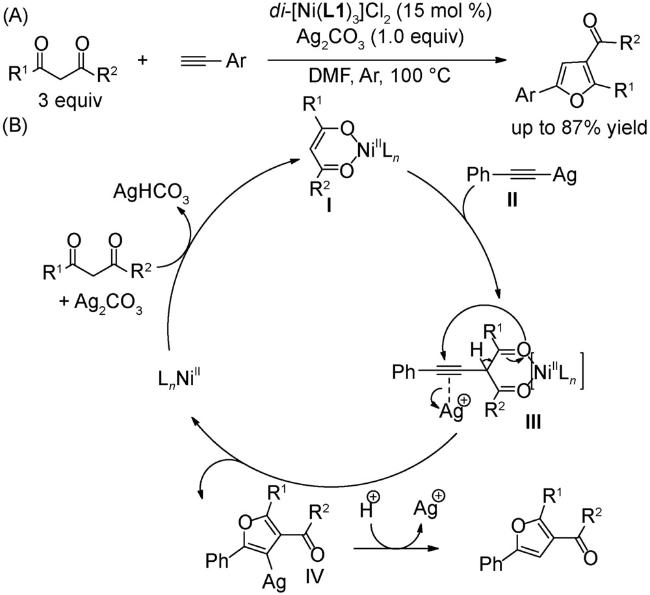
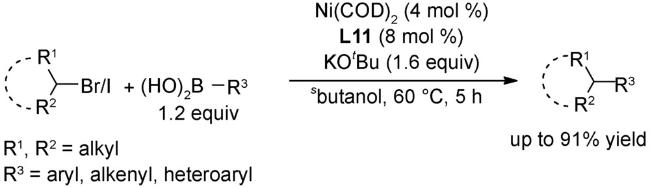
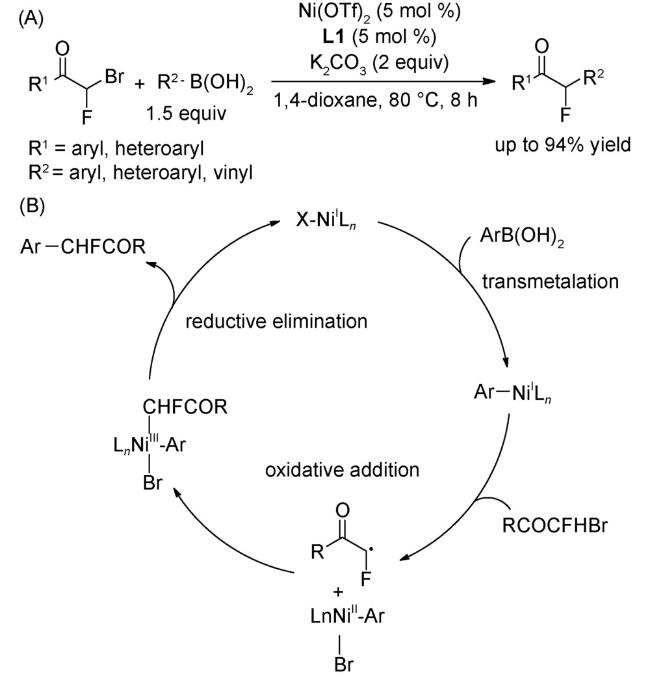


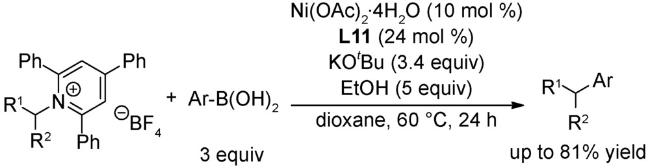
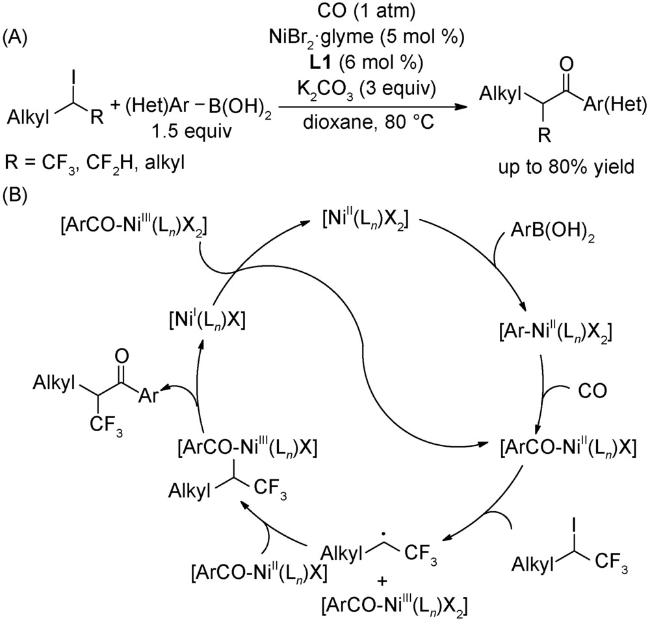
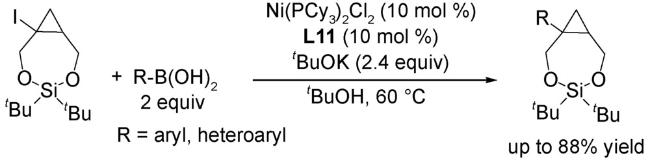
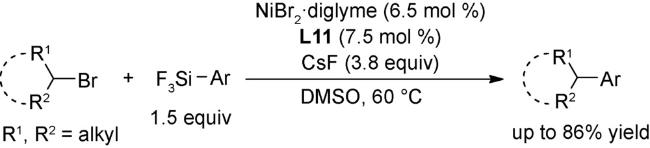
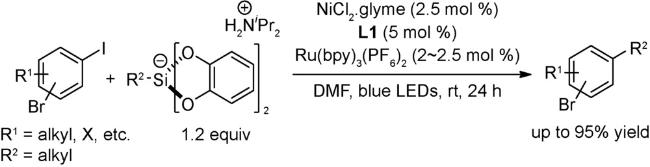
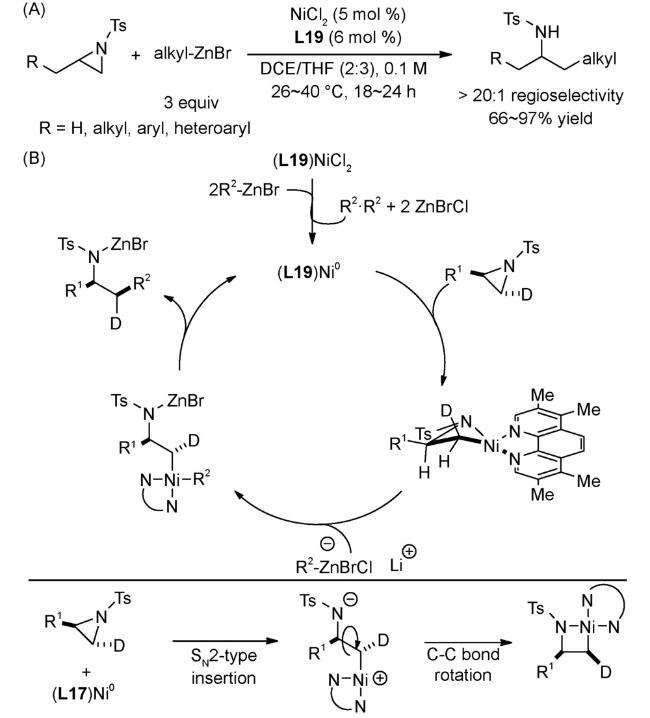
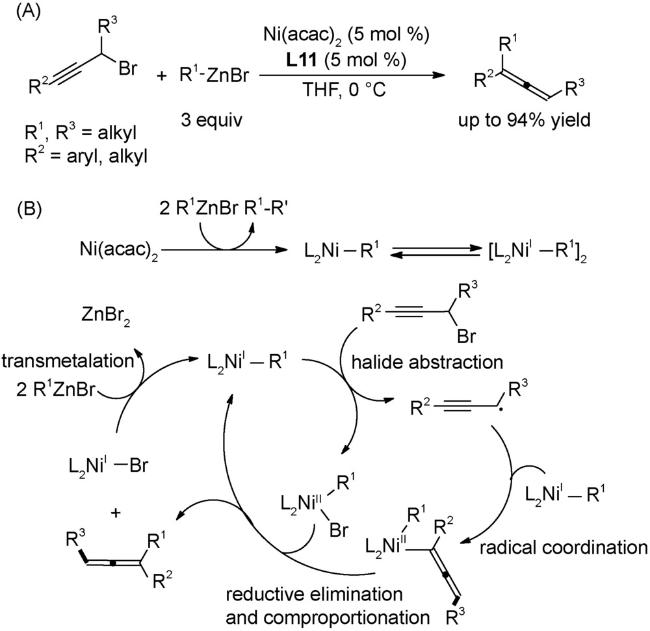
4.2 还原偶联反应
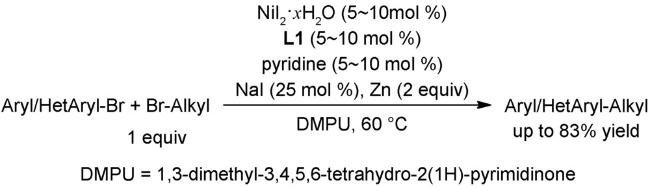
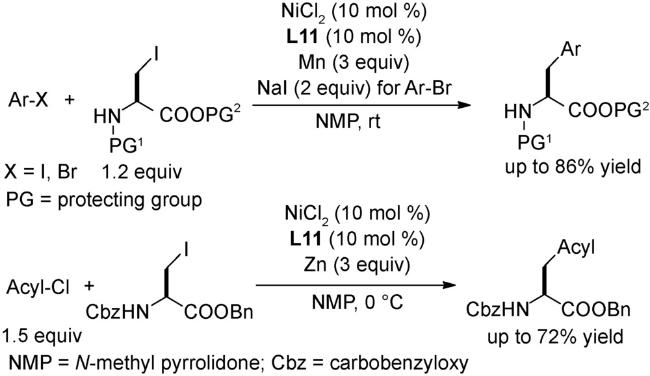
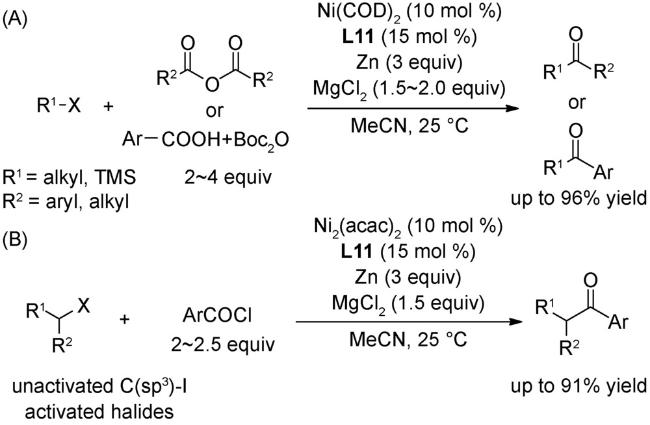
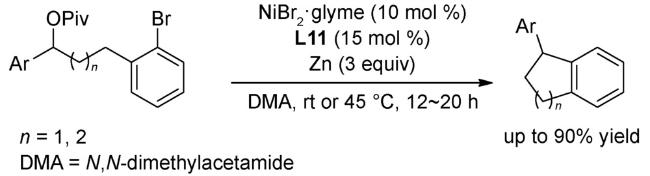

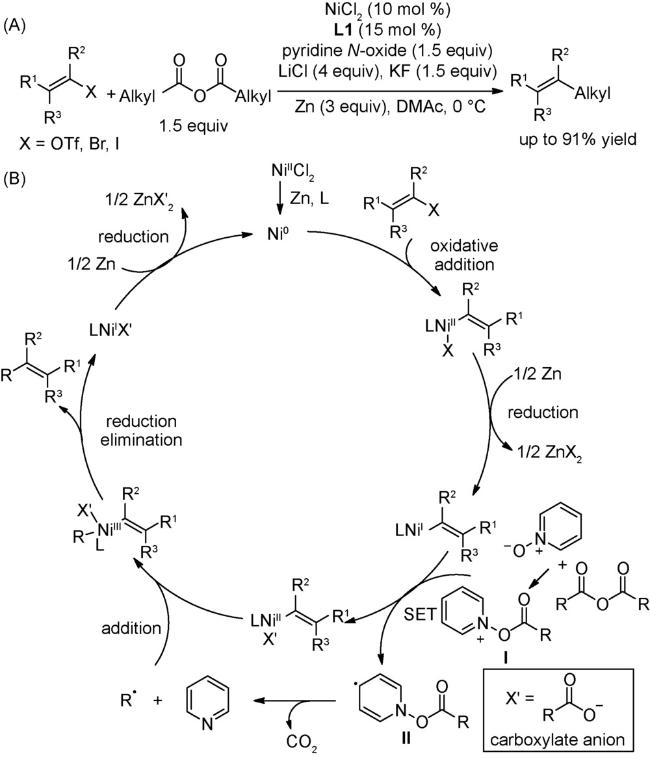
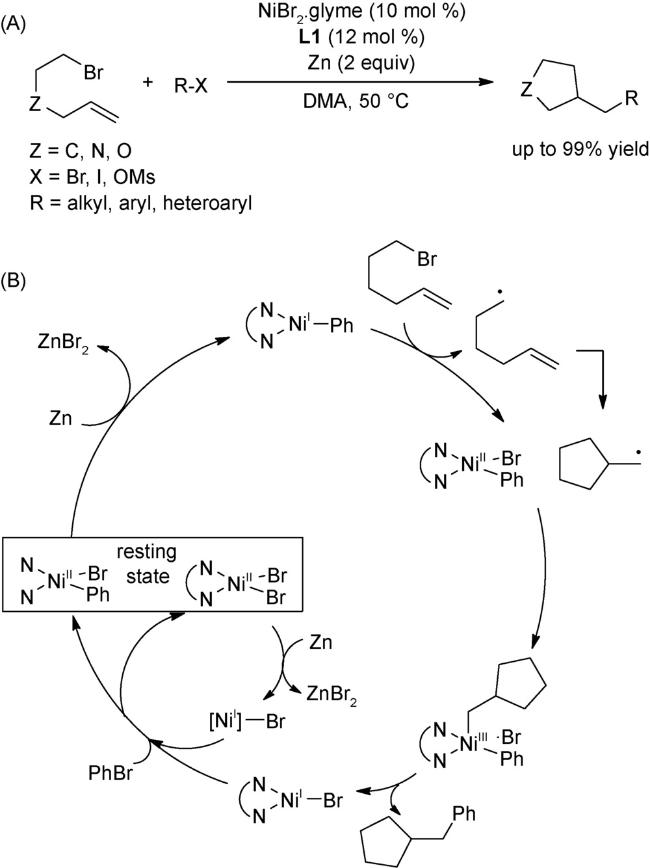
图式67 (A)镍催化的烷基亲电试剂和芳基溴代物的链行走还原交叉偶联反应[108];(B)可见光与镍共同催化的链行走还原交叉偶联反应[109];(C)芳基卤代物和烷基溴的电化学还原链行走交叉偶联反应[110]Scheme 67 (A) Nickel-catalyzed reductive relay cross-coupling of alkyl bromides and aryl bromides[108];(B) Photochemical nickel-catalyzed reductive migratory cross-coupling of alkyl bromides with aryl bromides[109];(C) Electrochemical reductive relay cross-coupling of alkyl halides to aryl halides[110] |
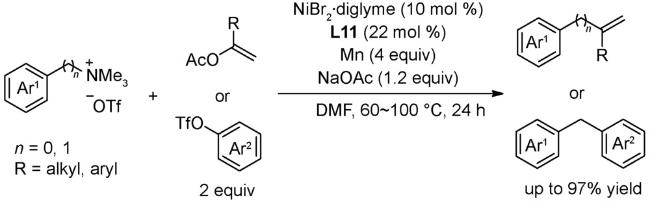
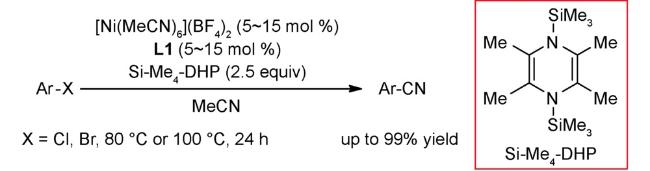
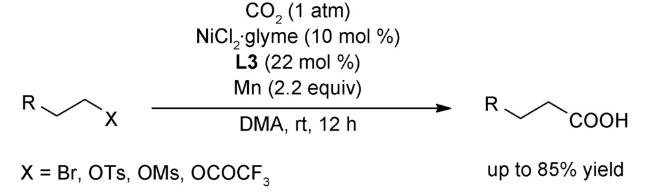
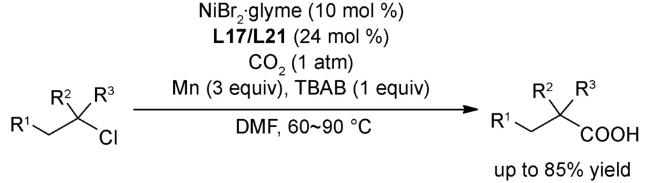
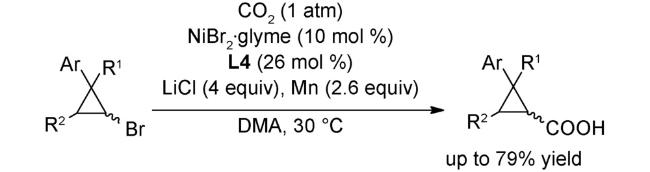
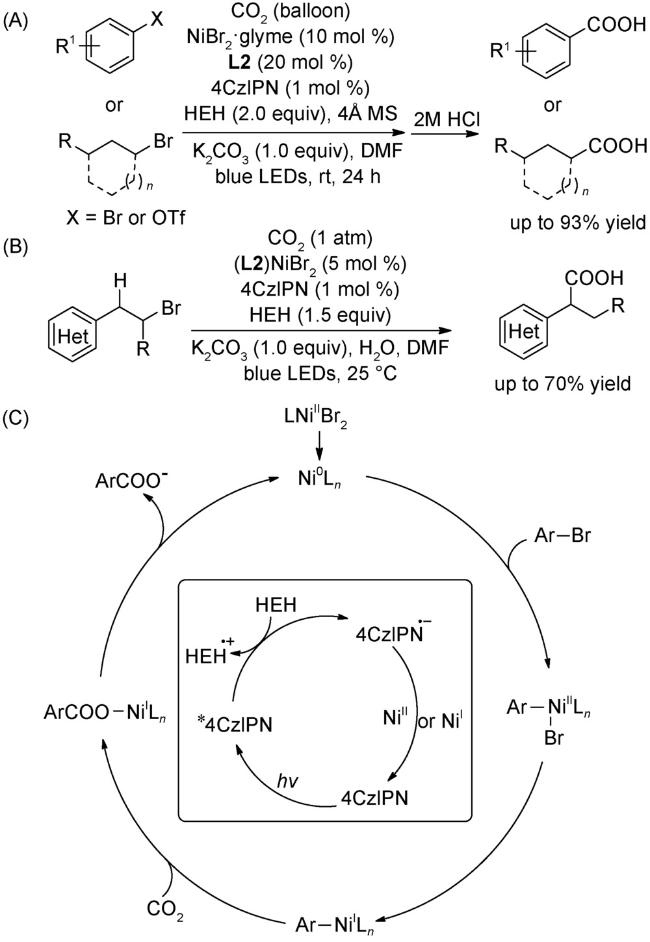
图式74 (A)光氧化还原与镍催化结合的溴代物与CO2的羧化反应[117];(B)光氧化还原与镍催化结合的远程C(sp3)-H键的羧化反应[118];(C)式(A)可能的机理[117]Scheme 74 (A) Carboxylation of aromatic and aliphatic bromides with CO2 by dual visible-light-nickel catalysis[117];(B) Remote C(sp3)-H carboxylation enabled by the merger of photoredox and nickel catalysis[118];(C) Proposed mechanism of (A)[117] |
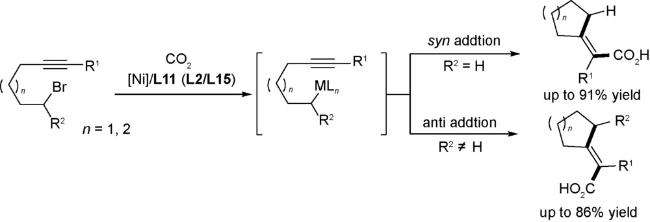
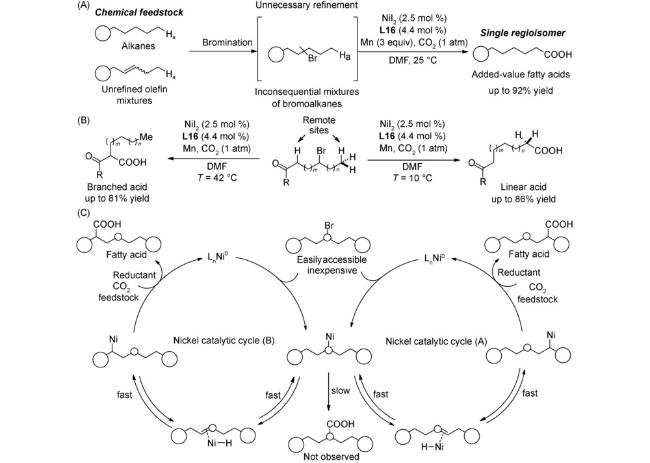
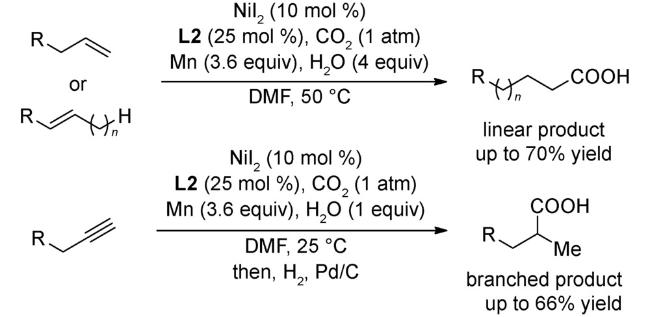
图式76 (A)经串联溴化/羧化反应将生物质原料直接催化转化为单一脂肪酸类化合物[120];(B)未活化烷基溴代物的远端C(sp3)-H键处的选择性羧化[120];(C)可能的机理[120]Scheme 76 (A) Application to the direct catalytic conversion of biomass-derived feedstocks into single fatty acids via a tandem bromination/carboxylation process[120];(B) Switchable site-selective carboxylation of unactivated alkyl bromides at remote C(sp3)-H sites[120];(C)Proposed mechanism[120] |
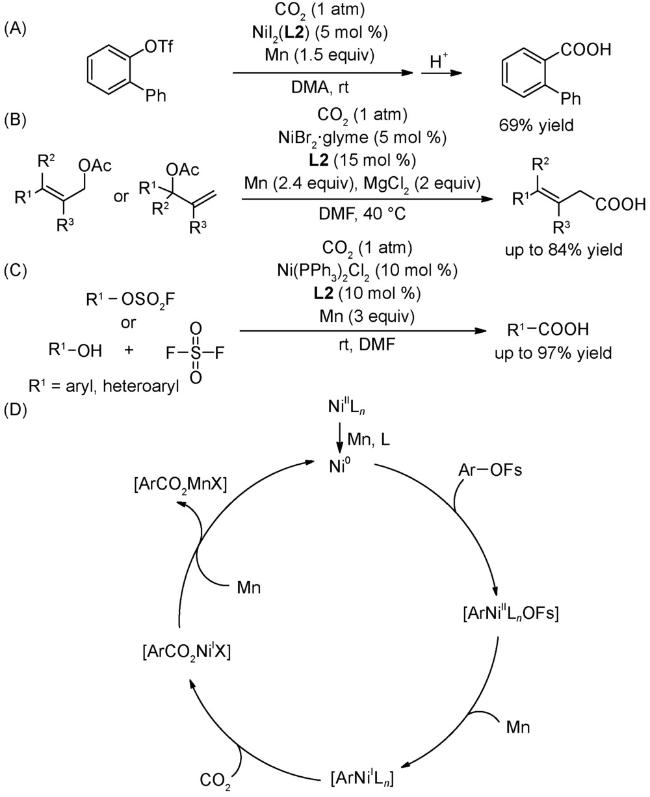
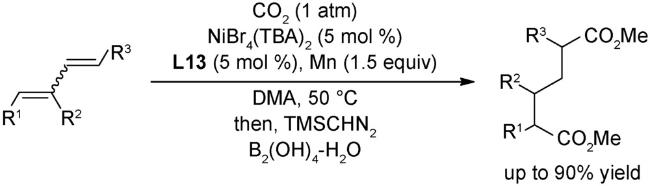
图式77 (A)烯基/大位阻的芳基三氟甲磺酸酯与CO2的羧化反应[70];(B)烯丙基酯与CO2的还原羧基化反应[121];(C)(杂)芳基氟磺酸酯与CO2的还原羧化反应[122];(D)式(C)可能的机理[122]Scheme 77 (A) Carboxylation of alkenyl and sterically hindered aryl triflates utilizing CO2[70];(B) Reductive carboxylation of allyl esters with C[122];(C) Carboxylation of aryl and heteroaryl fluorosulfates with CO2[122];(D) Proposed mechanism of (C)[122] |
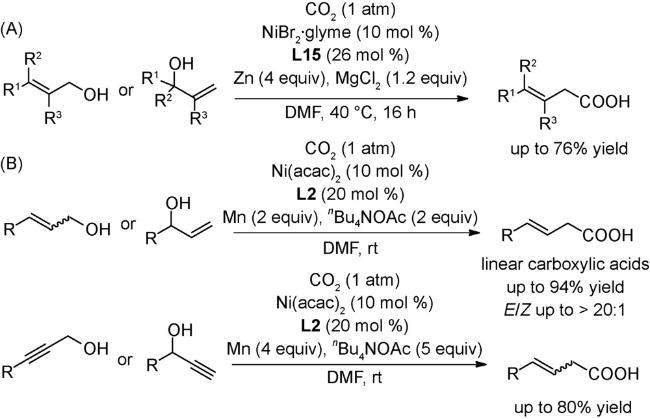
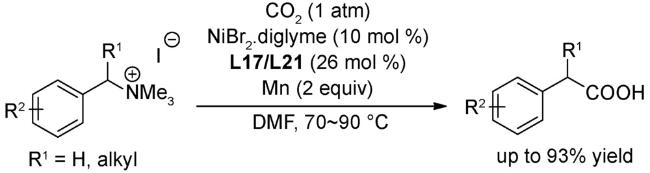
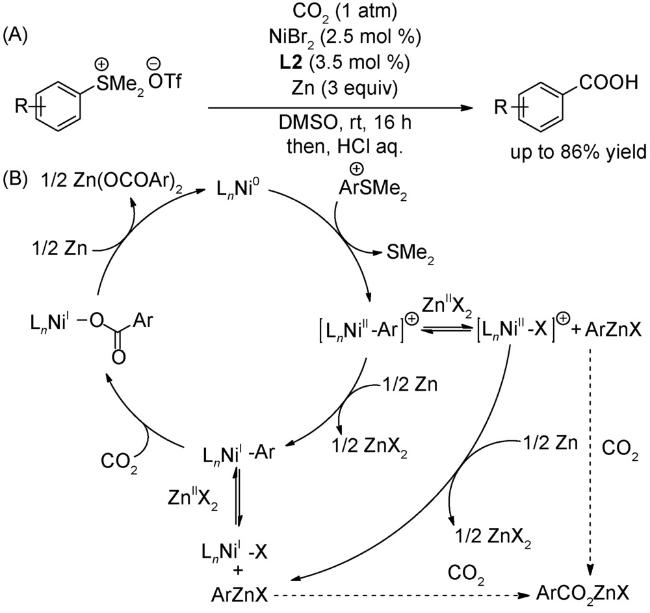
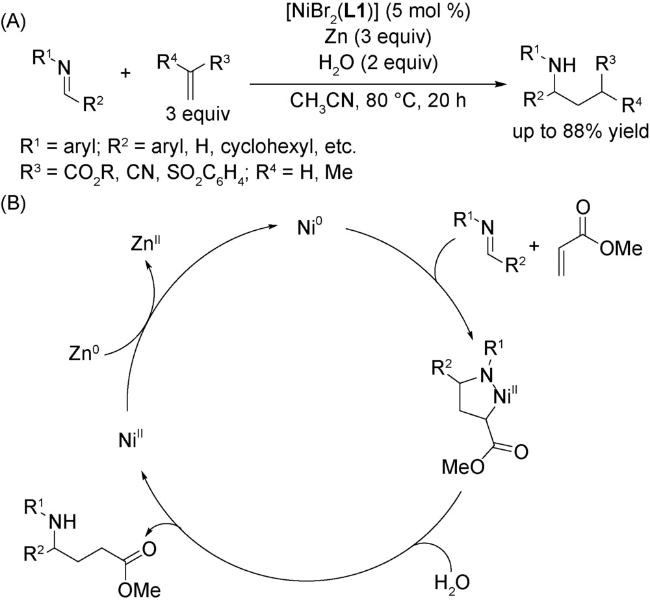
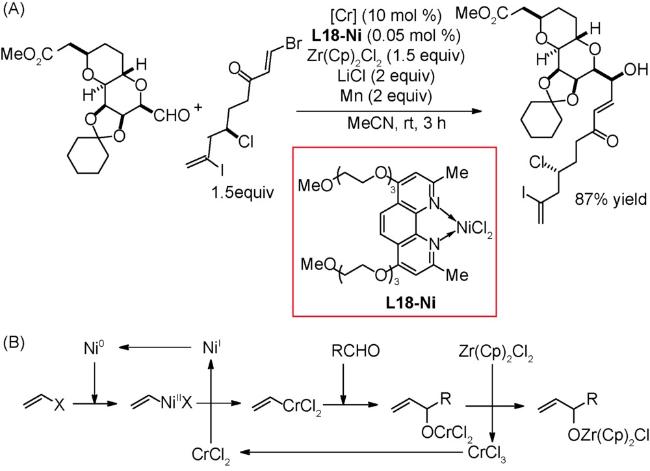
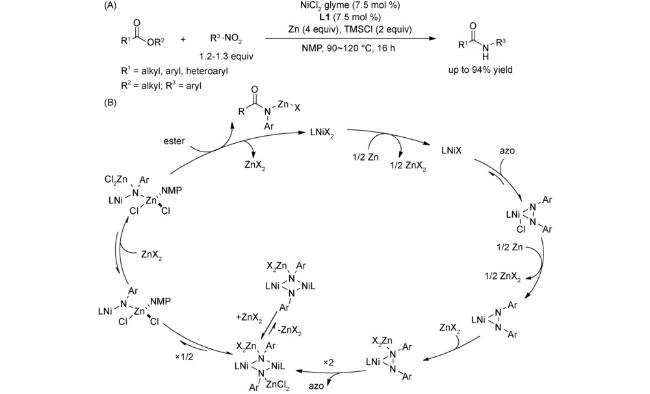
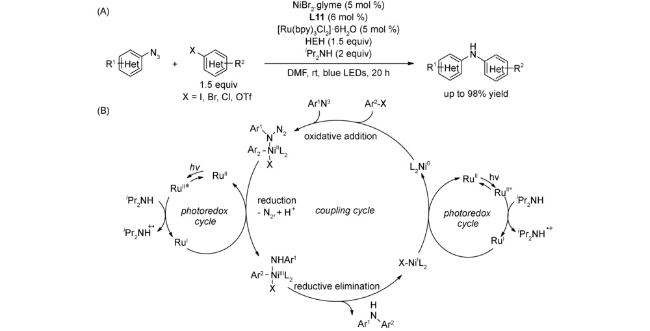
4.3 氧化偶联
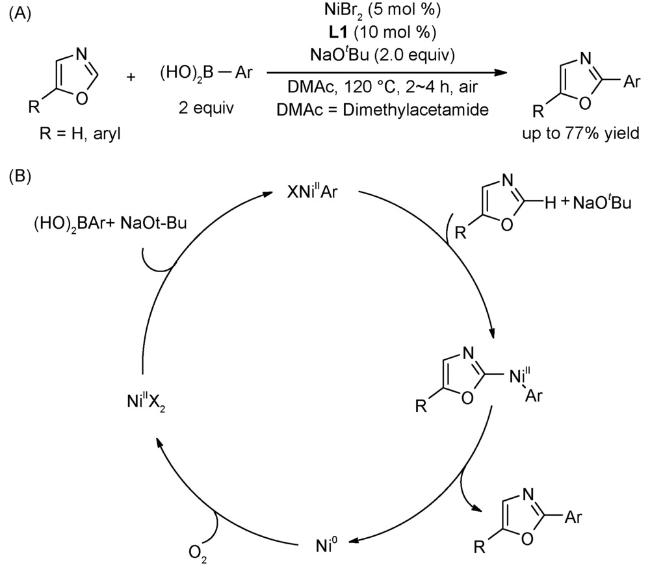
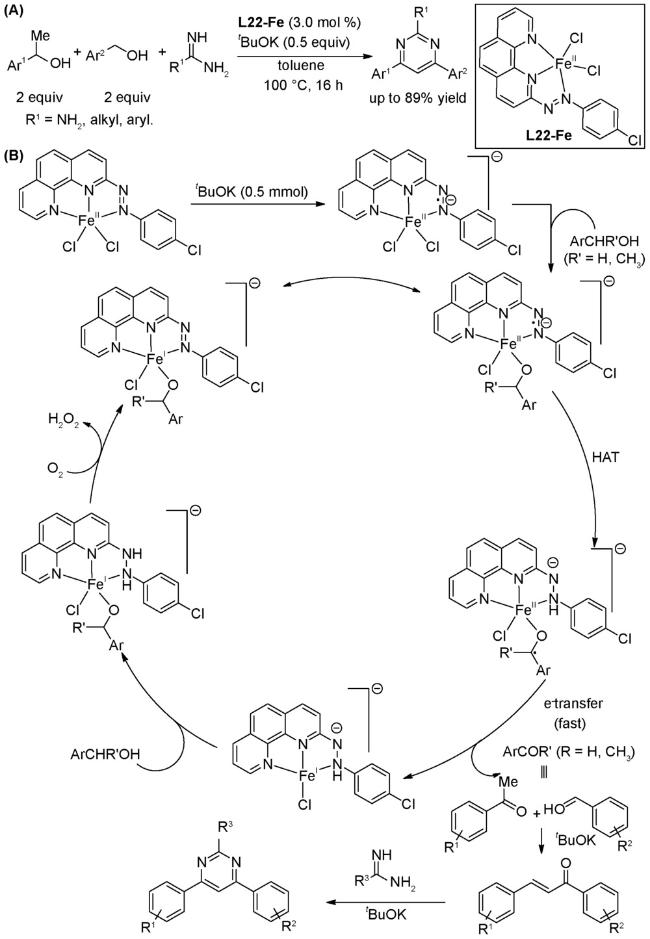
4.4 借氢反应
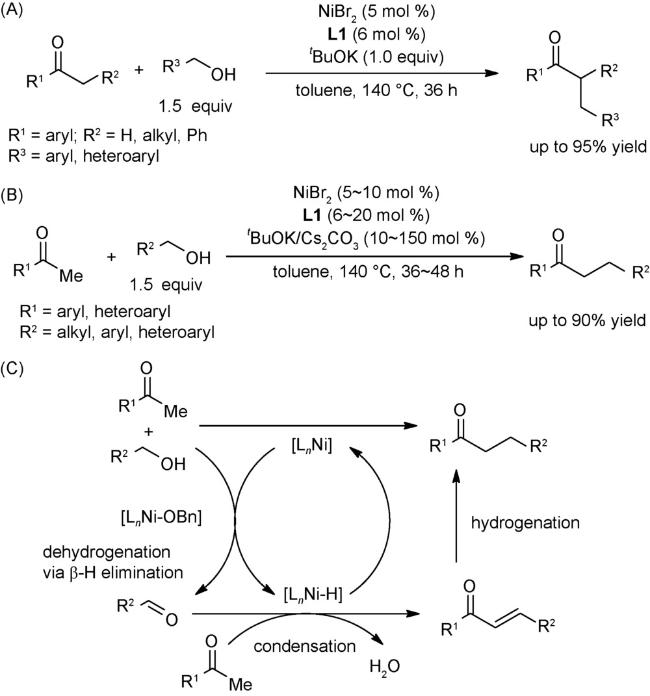
图式86 (A)酮与醇的α-烷基化反应合成支链偕二烷基酮[133];(B)甲基酮与醇的α-烷基化反应合成线性单取代酮[134];(C)可能的机理[134]Scheme 86 (A) α-Alkylation of ketones with alcohols to prepare branched gem-bis(alkyl) ketones[133];(B) α-Alkylation of methyl ketones with alcohols for the synthesis of monoselective linear ketones[134];(C) Proposed mechanism of (B)[134] |
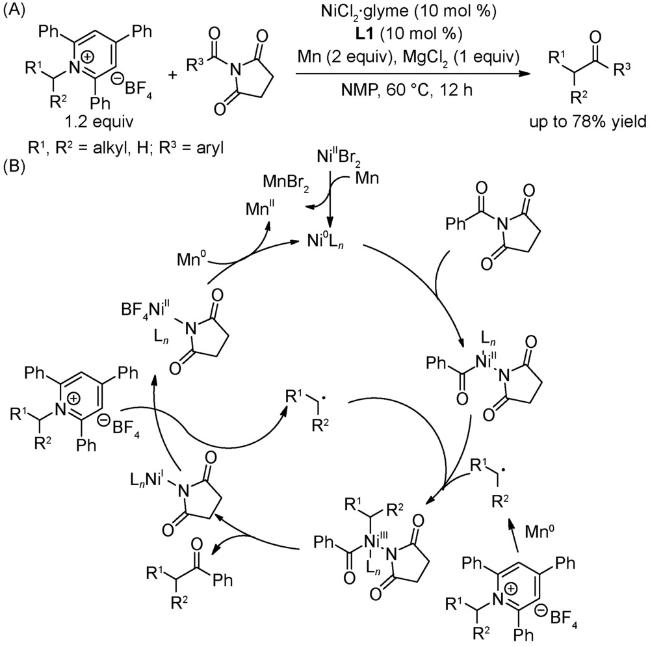
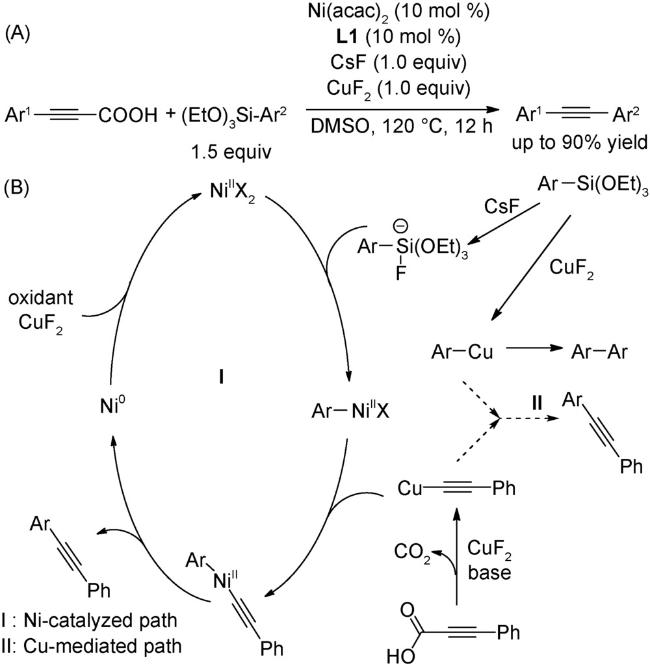
 C键被Ni—H物种还原,最后在碱作用下,发生消除反应得到最终产物。
C键被Ni—H物种还原,最后在碱作用下,发生消除反应得到最终产物。

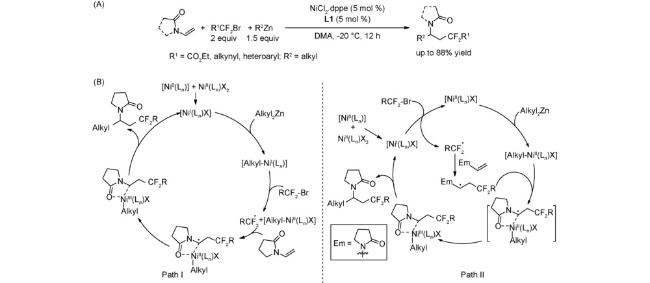

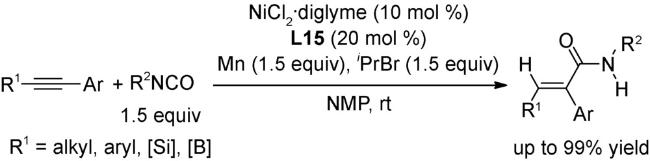
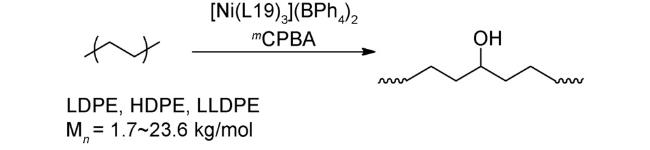
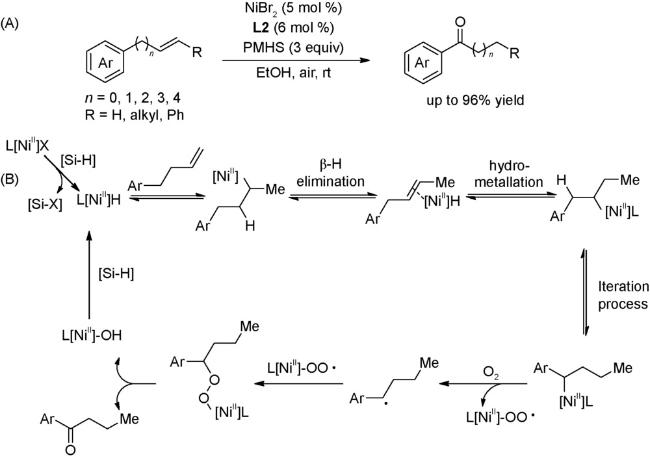
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}