





1 引言
分子管结构是合成化学基础研究的重要目标[1,2,3],理论上它们还具有很多潜在的功能,如包结、输送和传导等,碳纳米管是最具代表性的例子[4,5,6]。但碳纳米管及其衍生物一般没有溶解性,其层数、孔径大小和长度及结构修饰等都极具挑战性,并且修饰后的结构常常表现出不同于修饰前结构的性质特征。从分子科学的角度看,大环分子如环肽等可以通过氢键等分子间相互作用驱动,形成堆积管结构[7]。在膜内等受限的屏蔽溶剂的环境中,化学家可以通过氢键或疏水作用驱动不同的大环堆积形成通道。但不同大环分子间的相互作用不能产生协同作用,因此在溶液中一般难以形成这类管状结构。通过线性模板把环糊精等大环串联在一起,再通过共价键连接相邻分子,可以形成稳定的大分子管[8]。但这些管结构内径受限于大环单元,不具备可调控性。
足够长的线性大分子、高分子或超分子理论上可以通过折叠形成弹簧型的管型结构[3,9]。多肽和蛋白质的α-螺旋是最重要的二级结构之一,但不能形成可利用的螺旋内穴。很多基于非芳香单元构建的非天然螺旋系列,如聚异酸[10,11,12]、β-螺旋和γ-螺旋等[13]制备和功能研究也都取得了很多进展,但这些非天然螺旋结构也都不形成空穴结构。芳香分子可以通过分子内氢键和疏溶剂作用等非共价键相互作用驱动形成螺旋二级结构[14,15,16,17,18,19,20,21,22,23,24,25,26]。这类芳香螺旋结构的重复单元具有结构多样性,很多此类结构可以形成较大的内穴,其内径也相对固定,螺旋管的深度或长度可以通过控制分子链的长度调控。通过单体聚合或短链螺旋体进一步的头-尾组装,还可以构建具有较长深度的高分子和超分子螺旋管。本文总结这类管状结构的合成或组装及其在分子识别和包结、手性诱导、跨膜通道构建及反应加速和催化等方面的应用。
2 芳香寡聚螺旋管
2.1 芳香酰胺和酰肼构筑基元
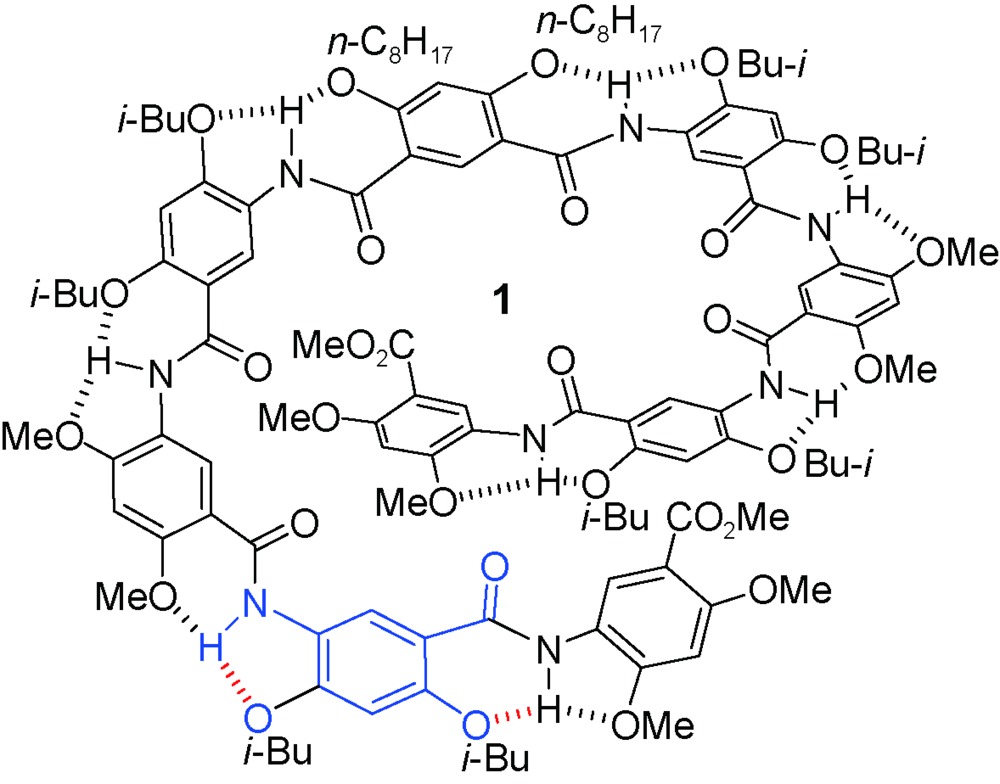
将刚性的芳香砌块用酰胺键连接,可以构建类似天然脂肪肽链一级结构的线性分子。分子内的氢键和疏溶剂作用等可以诱导相应的线性分子定向弯曲,而足够长的线性分子则可以弯曲形成螺旋结构[21]。各类芳香酰胺、酰肼和杂环都能够作为芳香重复单元,通过合理设计的氢键构筑不同类型的寡聚螺旋管[3,19],其形状和尺寸可以由芳香砌块来调节[27]。取代基位置差异以及不同取代位置的单体数量的排列编号可以调控螺旋折叠体的内径尺寸,最大可达10 Å以上。大的空腔能通过分子间非共价作用包结离子或小分子化合物。例如,芳香酰胺九聚体1由两条5-氨基-2,4-二烷氧基苯甲酸四聚体与2,4-二烷氧基-1,5-苯二甲酸缩合得到,侧链上烷氧基的氧原子作为良好的氢键受体与酰胺NH形成分子内N—H…O三中心氢键,使得整个酰胺寡聚体弯曲形成螺旋结构,随着链长增长,上下重叠的芳香单元之间产生堆积作用,进一步稳定了螺旋二级结构。
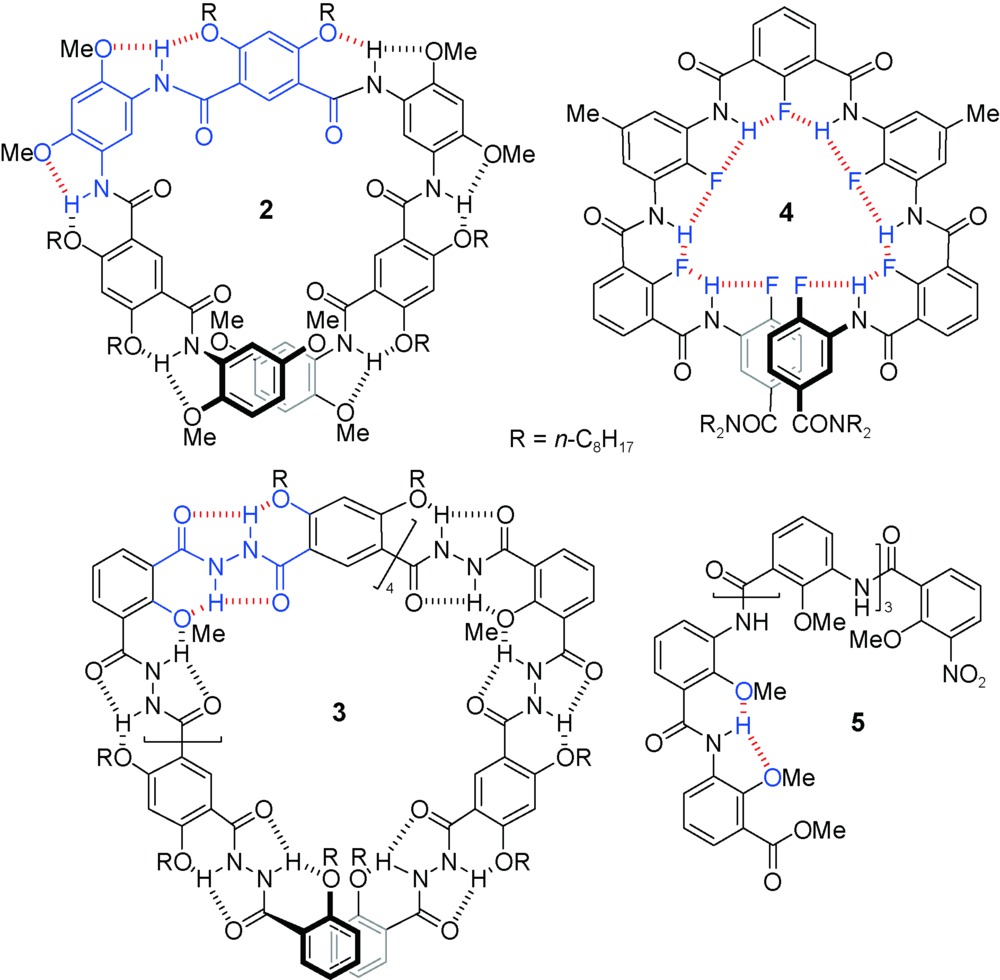
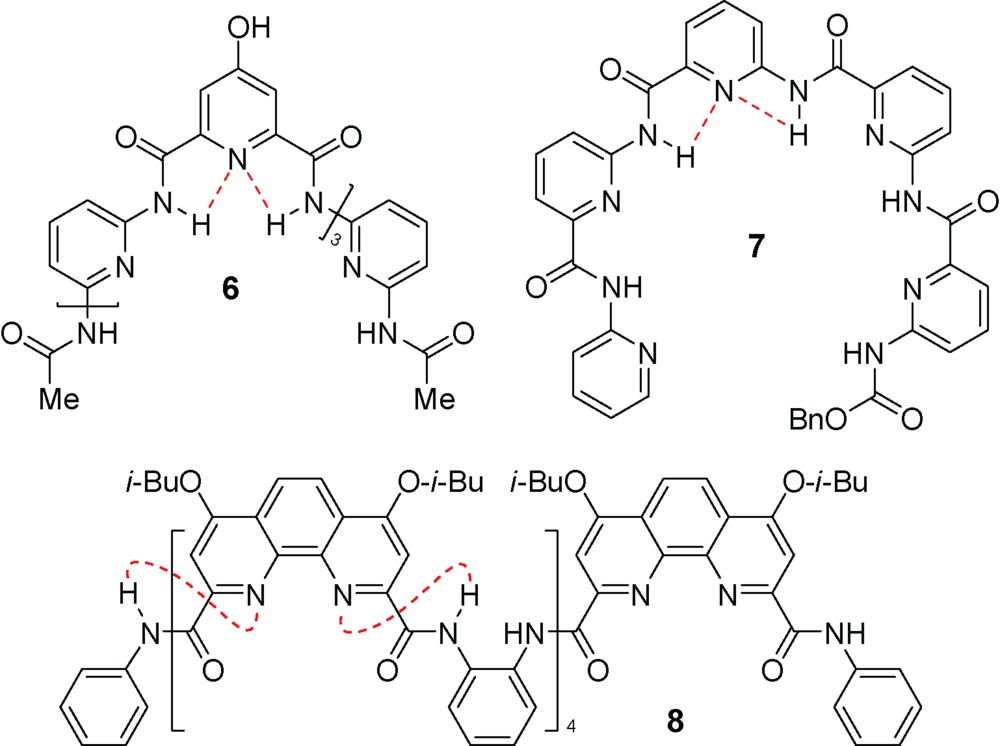
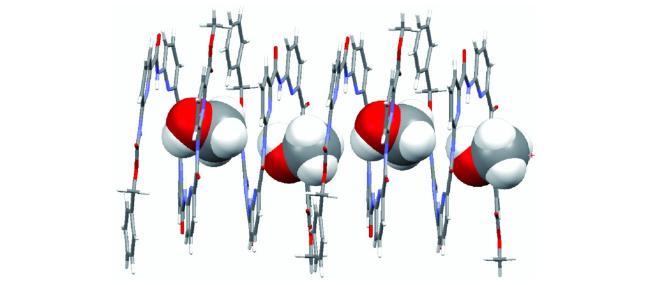
寡聚体2和3具有连续的分子内氢键,酰胺寡聚体的分子内氢键位于螺旋管的外部,而酰肼寡聚体的分子内氢键同时处于螺旋管的内部和外部。两类寡聚体的空腔尺寸较大,内径达到9~11 Å,足以容纳一些小分子化合物。酰胺羰基都朝向螺旋管的内部,通过分子间O—H…O(  C)氢键将烷基化的手性单糖、双糖等稳定在螺旋体空腔[28,29],手性单糖还进一步诱导产生了手性螺旋。寡聚体3有13个重复单元,能够形成约2.5圈的螺旋构象,较深的内穴使其与单糖的结合作用也比较强。较短的寡聚体4和5所形成的内穴则比较浅,并且控制分子构象的分子内氢键在空腔内部,内穴尺寸较小,然而这两个寡聚体仍能够作为主体分子结合铵盐[30,31]。寡聚螺旋管的内穴深度由于芳香砌块重复单元数量的不同而不同。8-氨基喹啉-2-甲酸三聚体能够形成一圈螺旋,其34-聚体形成了长度达到5.1 nm的螺旋管[32]。在其N-端和C-端分别引入电子给体和电子受体实现了光诱导的电子转移。吡啶寡聚体7的晶体结构(图1)表明约4.3个重复单元即可以形成一圈螺旋,但其空腔较小,仅能容纳水分子[33,34]。引入邻二氮杂菲的寡聚体8没有形成酰胺基团参与的分子内氢键,而是与吡啶的氮原子形成了分子内五元环N—H…N氢键,通过这种分子内氢键作用使整个分子发生折叠,形成了一个没有可利用空腔的螺旋结构。晶体结构显示(图2),所有酰胺的羰基氧原子朝向螺旋的外部,水分子通过分子间氢键作用占据了螺旋体的空腔[35]。吡啶、喹啉、吡啶并喹啉等芳杂环酰胺单元通过合理的序列设计形成螺旋结构,这些螺旋体与其他分子可以组装形成分子梭等主客体超分子结构[36,37,38]。2,6-二氨基吡啶与2,6-吡啶二酸的缩合寡聚体、7-氨基8-氟-2-喹啉酸的寡聚体等也形成螺旋结构,并进一步组装为双螺旋或多重螺旋结构,但这些螺旋结构的空腔尺寸较小,无法容纳客体分子[26,39,40]。
C)氢键将烷基化的手性单糖、双糖等稳定在螺旋体空腔[28,29],手性单糖还进一步诱导产生了手性螺旋。寡聚体3有13个重复单元,能够形成约2.5圈的螺旋构象,较深的内穴使其与单糖的结合作用也比较强。较短的寡聚体4和5所形成的内穴则比较浅,并且控制分子构象的分子内氢键在空腔内部,内穴尺寸较小,然而这两个寡聚体仍能够作为主体分子结合铵盐[30,31]。寡聚螺旋管的内穴深度由于芳香砌块重复单元数量的不同而不同。8-氨基喹啉-2-甲酸三聚体能够形成一圈螺旋,其34-聚体形成了长度达到5.1 nm的螺旋管[32]。在其N-端和C-端分别引入电子给体和电子受体实现了光诱导的电子转移。吡啶寡聚体7的晶体结构(图1)表明约4.3个重复单元即可以形成一圈螺旋,但其空腔较小,仅能容纳水分子[33,34]。引入邻二氮杂菲的寡聚体8没有形成酰胺基团参与的分子内氢键,而是与吡啶的氮原子形成了分子内五元环N—H…N氢键,通过这种分子内氢键作用使整个分子发生折叠,形成了一个没有可利用空腔的螺旋结构。晶体结构显示(图2),所有酰胺的羰基氧原子朝向螺旋的外部,水分子通过分子间氢键作用占据了螺旋体的空腔[35]。吡啶、喹啉、吡啶并喹啉等芳杂环酰胺单元通过合理的序列设计形成螺旋结构,这些螺旋体与其他分子可以组装形成分子梭等主客体超分子结构[36,37,38]。2,6-二氨基吡啶与2,6-吡啶二酸的缩合寡聚体、7-氨基8-氟-2-喹啉酸的寡聚体等也形成螺旋结构,并进一步组装为双螺旋或多重螺旋结构,但这些螺旋结构的空腔尺寸较小,无法容纳客体分子[26,39,40]。
C)氢键将烷基化的手性单糖、双糖等稳定在螺旋体空腔[28,29],手性单糖还进一步诱导产生了手性螺旋。寡聚体3有13个重复单元,能够形成约2.5圈的螺旋构象,较深的内穴使其与单糖的结合作用也比较强。较短的寡聚体4和5所形成的内穴则比较浅,并且控制分子构象的分子内氢键在空腔内部,内穴尺寸较小,然而这两个寡聚体仍能够作为主体分子结合铵盐[30,31]。寡聚螺旋管的内穴深度由于芳香砌块重复单元数量的不同而不同。8-氨基喹啉-2-甲酸三聚体能够形成一圈螺旋,其34-聚体形成了长度达到5.1 nm的螺旋管[32]。在其N-端和C-端分别引入电子给体和电子受体实现了光诱导的电子转移。吡啶寡聚体7的晶体结构(图1)表明约4.3个重复单元即可以形成一圈螺旋,但其空腔较小,仅能容纳水分子[33,34]。引入邻二氮杂菲的寡聚体8没有形成酰胺基团参与的分子内氢键,而是与吡啶的氮原子形成了分子内五元环N—H…N氢键,通过这种分子内氢键作用使整个分子发生折叠,形成了一个没有可利用空腔的螺旋结构。晶体结构显示(图2),所有酰胺的羰基氧原子朝向螺旋的外部,水分子通过分子间氢键作用占据了螺旋体的空腔[35]。吡啶、喹啉、吡啶并喹啉等芳杂环酰胺单元通过合理的序列设计形成螺旋结构,这些螺旋体与其他分子可以组装形成分子梭等主客体超分子结构[36,37,38]。2,6-二氨基吡啶与2,6-吡啶二酸的缩合寡聚体、7-氨基8-氟-2-喹啉酸的寡聚体等也形成螺旋结构,并进一步组装为双螺旋或多重螺旋结构,但这些螺旋结构的空腔尺寸较小,无法容纳客体分子[26,39,40]。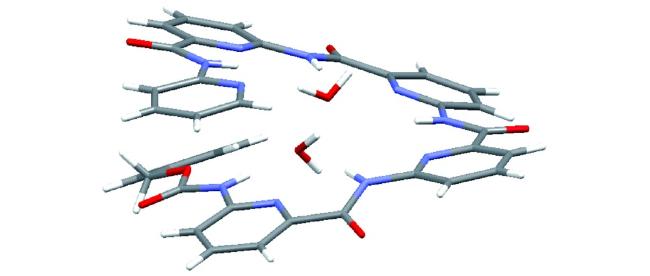
2.2 芳香三氮唑构筑基元
利用特定的杂环序列形成反式构象的特点(“螺旋密码子”),设计嵌入这类单元的长链分子,也可以构建螺旋结构[41]。1,2,3-三唑非常易于通过点击反应制备[42,43,44,45,46],使其能够方便地被并入到这类序列中。不同于一般通过分子内氢键控制螺旋结构的芳酰胺寡聚体,并入苯和1,4-二取代-1,2,3-三唑/吡啶的九聚体9a和十七聚体9b在乙腈和水的混合溶剂中形成了非氢键驱动的螺旋结构。疏溶剂作用是这类寡聚体折叠的主要驱动力,足够长的寡聚体形成超过一圈的螺旋结构,则螺旋重叠的部分芳基之间形成π-π堆积稳定了螺旋二级结构[47]。较长的寡聚体9b由于手性侧链的诱导产生了螺旋手性。有趣的是螺旋的手性会由于KCl、KBr或HCl的加入发生翻转,而加入KF几乎没有变化。这说明识别作用和卤素负离子的尺寸及体系的酸性有很大关系,分子识别的作用位点在缩乙二醇侧链。
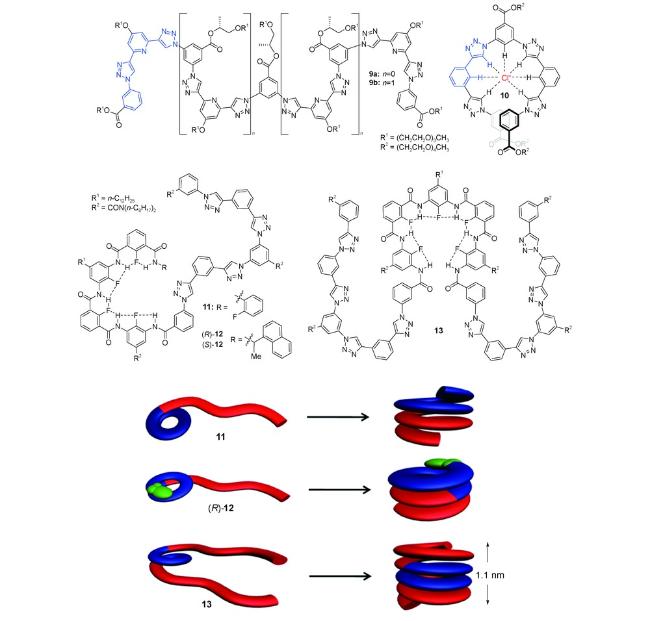
1,2,3-三唑的偶极矩为1.2 D,其5-位氢原子具有一定酸性,这个结构特点使其与酰胺键类似[48]。1,4-二芳基-1,2,3-三唑寡聚体有柔性骨架,寡聚体10与卤素负离子形成多价分子间氢键(C—H…Cl-)使其形成螺旋结构,5-位的氢原子朝向螺旋内部[49,50]。引入适当修饰的苯环或吡啶盐的类似寡聚体也能形成折叠构象,并且能够在有机溶剂或水溶液中与卤素负离子表现出良好的结合能力[51,52]。受分子内N—H…F氢键作用驱动,寡聚体4形成螺旋二级结构[30],将超过4个重复单元的氟代芳酰胺片段与柔性1,4-二芳基-1,2,3-三唑寡聚体结合构建的寡聚体11~13在苯或正己烷等溶剂中形成了稳定构象[53]。氟代芳酰胺片段形成的固有螺旋结构诱导1,4-二芳基-1,2,3-三唑寡聚体片段产生折叠,不论固有螺旋片段位于寡聚体端位的11和12,还是位于中间位置的寡聚体13,均能诱导1,4-二芳基-1,2,3-三唑寡聚体部分形成螺旋构象,而在氯仿或二氯甲烷溶剂中未观察到这一现象,说明这种诱导效应可归因于疏溶剂作用促进的分子内芳环的层间堆积。当寡聚体12固有螺旋的N-端引进手性单元,产生手性固有螺旋,进一步诱导没有手性的1,4-二芳基-1,2,3-三唑寡聚体形成了手性螺旋,也即螺旋可以传递,螺旋的手性也可以传递。螺旋诱导螺旋的示意图如图3所示,寡聚体13形成的螺旋管深度约为1.1 nm。

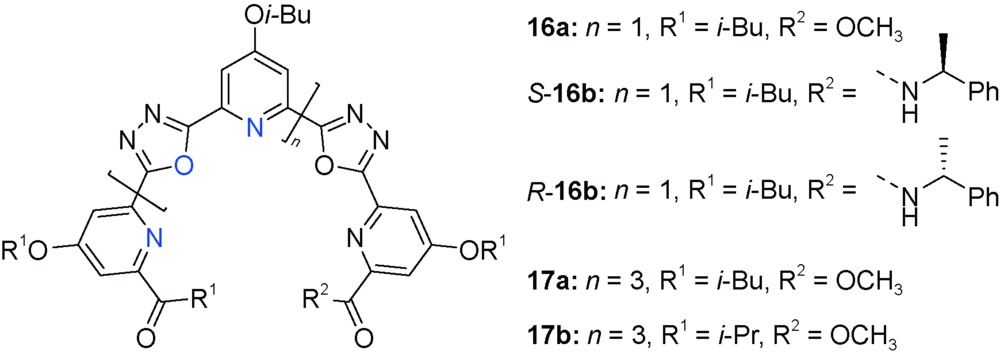
用1,3,4-口恶二唑替换1,2,3-三唑构建的吡啶/口恶二唑寡聚体展现出与吡啶/三唑寡聚体类似的螺旋构象,口恶二唑的2个吡啶取代基采取顺式构象,吡啶氮原子与口恶二唑的氧原子均朝向螺旋结构的内部[56]。手性相反的五聚体S-16b和R-16b表现出互为镜像的Cotton效应,说明其形成手性相反的螺旋体。五聚体16a和九聚体17a,17b与K+选择性结合,在CH3CN/H2O(9∶1) 中结合常数分别为614±18(16a/K+=1∶1),2178±87(17a/K+=2∶3) 和2351±82(17b/K+=2∶3)。五聚体16a和九聚体17b嵌膜形成人工通道,九聚体形成的跨膜通道对K+/Na+选择性相差22.5倍,说明其螺旋结构产生连续的结合位点利于K+识别,从而实现了选择性人工跨膜离子传导。
2.3 间-苯乙炔和杂环类似物构筑基元
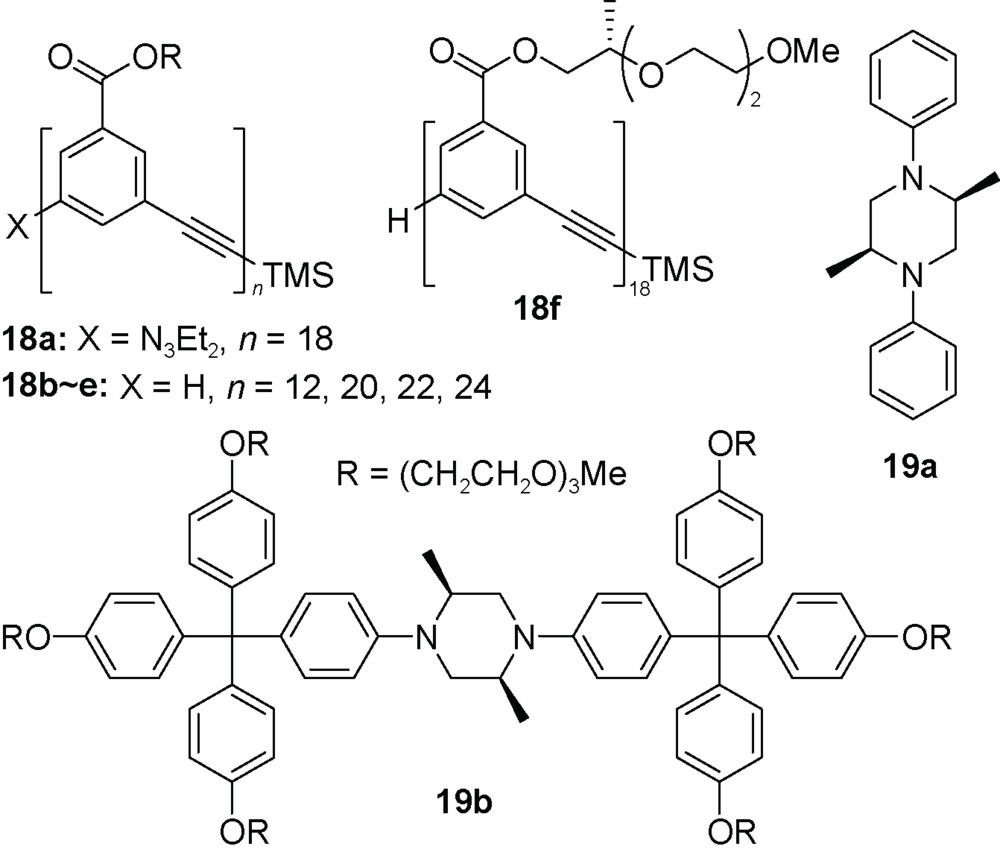
不同于氢键作用驱动线性分子形成螺旋结构的酰胺或三唑类骨架,间-苯乙炔(mPE)作为重复单元的寡聚体不形成分子内或者分子间氢键,这类寡聚体折叠的主要驱动力是疏溶剂作用[57]。在乙腈等极性有机溶剂中,有10个以上间-苯乙炔重复单元的寡聚体形成有序构象,十八聚体18a形成稳定的螺旋结构。约5个间-苯乙炔单元构成一圈螺旋,所以10个单元形成2圈螺旋,产生的π-π堆积稳定了螺旋构象,十八聚体形成4圈螺旋结构。芳香堆积作用在氯仿等低极性有机溶剂中非常弱,这也是间-苯乙炔寡聚体在低极性溶剂中不形成螺旋构象的重要原因。紫外和核磁共振氢谱等研究手段均发现,无论是短的间-苯乙炔寡聚体还是长的寡聚体,在氯仿中均呈现为无规卷曲构象。间-苯乙炔螺旋管的内径尺寸为8.7 Å,空腔可以包结疏水性小分子化合物。在40%乙腈水溶液中,间-苯乙炔十二聚体18b能够与α-蒎烯形成1∶1复合物,并且分别与(-)-α-蒎烯和(+)-α-蒎烯结合表现互为镜像的Cotton效应[58]。较长的寡聚体18c,d能够与棒状的19a和19b相结合[59]。棒状的哌嗪衍生物19a直径约为6.6 Å,两端引入四苯甲烷衍生物,端位的大基团尺寸约为10.2 Å。在40%乙腈水溶液中,寡聚体18c,e(n=20, 22, 24) 分别与19a或19b形成1∶1的复合物。由于19b的哑铃状结构,这种结合过程很可能经过一种动力学过程,间-苯乙炔螺旋管首先发生解螺旋,再发生缠绕而恢复螺旋结构;由于端位大体积基团会阻碍螺旋结构的形成,二十四聚体18e与19b的结合要明显弱于18c或18d与19b的结合。在缩乙二醇侧链引入甲基,得到含手性侧链的间-苯乙炔寡聚体18f[60]。圆二色谱实验发现,手性十八聚体18f在氯仿溶剂中没有Cotton效应,说明寡聚体在氯仿中的构象是无序的。在乙腈中十八聚体有强的Cotton效应,说明其形成了手性螺旋结构,这种骨架的手性由侧链手性传递而来。
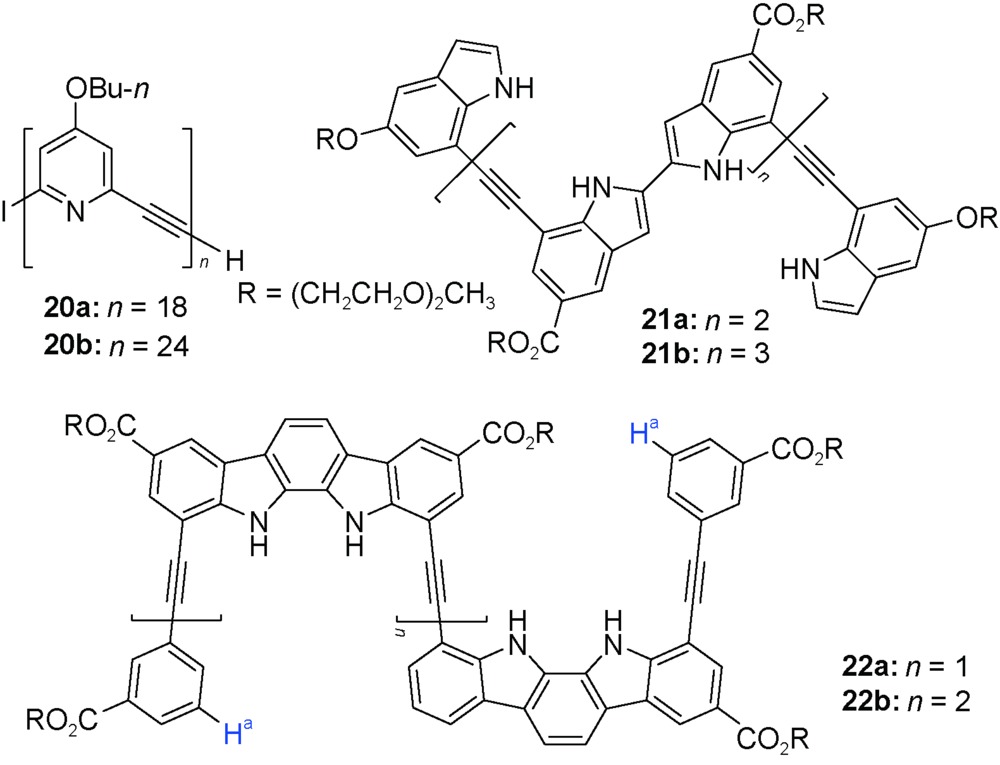
联吲哚在溶液中也倾向于采取s-trans构象。在聚吲哚21a和21b的乙腈溶液中加入四丁基氯化铵,聚吲哚的构象发生改变,通过与氯负离子形成1∶1的结合,表观结合常数分别达到1.2(± 0.1)×106 M-1(21a·Cl-) 和 > 107 M-1(21b·Cl-)。聚吲哚的构象转化为s-cis,从而形成了螺旋结构[62]。由于聚吲哚单元的尺寸远远大于吡啶,形成螺旋结构所需的重复单元数大大减少。相较于联吲哚,吲哚并咔唑基元的2个NH被固定在一侧,将其作为构建基元得到的聚乙炔吲哚并咔唑比聚吲哚更刚性[63]。在大极性的DMSO/MeOH(4∶1) 溶剂中,聚合物22a和22b与氯离子以1∶1结合,表观结合常数分别为560 M-1(22a·Cl-) 和36 800 M-1 (22b·Cl-)。进一步将酯基水解得到羧酸钠盐,其在水溶液中与氯负离子形成分子间氢键而折叠形成螺旋结构。计算表明,对于三聚体,除了6个吲哚并咔唑的NH参与形成分子间氢键,两个端基的苯基CHa也参与形成分子间C—H…Cl-氢键,并控制构象产生了T-型芳香堆积。在水溶液中,其结合卤素负离子的能力与有机相中顺序不同,表现出溶剂化效应对分子间氢键作用的影响。
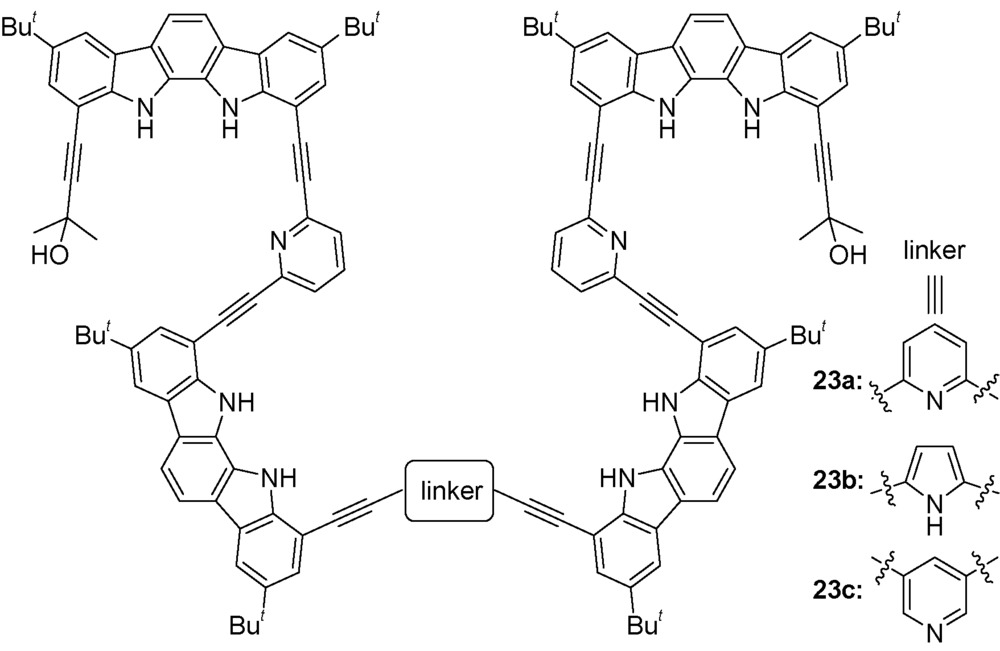
吲哚并咔唑和2,6-二取代吡啶组成的聚合物23a也能够在水溶液中形成螺旋结构,由于吲哚并咔唑的NH和吡啶的氮原子与溶剂水分子产生分子间氢键,在螺旋的空腔中形成了水线[64]。将位于线性聚合物中间位置的2,6-二取代吡啶置换为2,5-二取代吡咯或3,5-二取代吡啶,改变了空腔内部氢键给受体的排列,氢键受体(吡啶氮原子)转变为氢键给体(吡咯NH或吡啶C4H),从而破坏了水分子的氢键网络。单晶结构表明聚合物23b和23c的空腔内结合了2个磷酸二氢根。氢键给受体的调节改变了主客体分子间的作用,这对于深入认识酶的功能等生命过程提供了一种手段。
3 芳香聚合物螺旋管
除少数寡聚体可以用固相合成法[57],一般线性聚合物都通过多步合成来构建,合成过程费时费力。通过设计合适的双官能团底物,一步聚合可能得到大分子量线性聚合物,将为探索具有深内穴螺旋管的功能提供研究对象。长的螺旋管在结构上与相应寡聚体类似,但在功能方面有独特之处。解决聚合物在溶液中的溶解度是研究其溶液相行为所面临的首要问题之一。
3.1 芳香酰胺和类似物构筑基元
通过设计不同结构的芳香重复单元和控制聚合度可以调节所形成的螺旋管内径尺寸及其深度。碱金属或碱土金属盐作为模板剂可以促进大环酰胺或聚酰胺的合成[65,66],也可以通过破坏产物的分子间氢键来提高聚合物的溶解度,这为聚酰胺的合成提供了有效方法。以3-氨基-2-甲氧基-5-甲基苯甲酸24为单体,在N-甲基吡咯烷酮(NMP)溶剂中,PPh3为缩合剂,4% LiCl存在下聚合,得到了聚合物P25(Mn=3.75 kDa, PDI=1.72)[67]。聚合过程中有部分甲醚键发生了断裂。聚合物P25通过硫酸二甲酯甲基化,得到聚合物P26(Mn=2.43 kDa, PDI=1.47),在碱的作用下形成去质子化的聚合物P27(Mn=2.07 kDa, PDI=1.52)(图式1)。聚合物P26和P27的分子量要小于P25,说明其结构与寡聚体5相类似,通过分子内N—H…O氢键控制聚芳酰胺的构象形成螺旋结构,所形成的螺旋管有约三圈螺旋,深度1.1 nm[68]。在DMSO溶液中,手性α-甲基苯乙酸诱导聚合物P26产生手性螺旋,聚合物P27则受手性α-甲基苯乙胺的诱导产生手性螺旋。
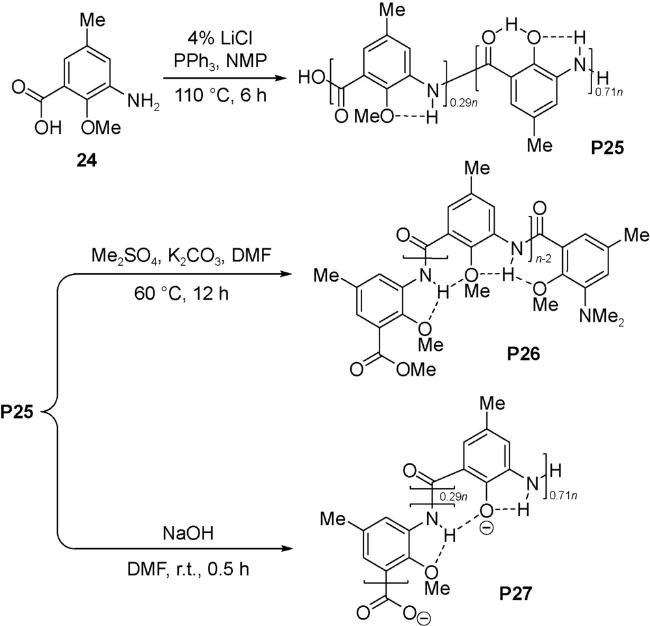
聚合物P28由间-苯二胺和间-苯二酰氯的衍生物缩合而成,在间-苯二酰氯单体中引入的烷氧基可以与酰胺键形成分子内N—H…O(R)氢键[69]。通过反复的良溶剂/不良溶剂沉淀法,聚合物P28被分为不同聚合度的三部分(PDI=1.02~1.03),Mn分别为10 200、18 600和35 100。与相应十二聚体模型化合物比较可知,三部分分别形成了约5、9和18圈螺旋。不论是P28还是P28a~c,聚合物在CH2Cl2、CHCl3、THF、DMF和DMA等多种有机溶剂中均有良好的溶解度。在CH2Cl2或CHCl3等低极性有机溶剂中的良好溶解度说明,聚合物采取螺旋构象而使侧链朝向螺旋外侧,避免了聚芳酰胺因分子间氢键作用聚集而不溶于有机溶剂,螺旋层间的两个酰胺侧链之间的N—H…O( C) 氢键则进一步稳定了螺旋构象。圆二色谱实验发现,随着链长逐渐增加,Cotton效应不断增强,说明侧链的手性会诱导螺旋管产生手性,并且具有协同效应。
C) 氢键则进一步稳定了螺旋构象。圆二色谱实验发现,随着链长逐渐增加,Cotton效应不断增强,说明侧链的手性会诱导螺旋管产生手性,并且具有协同效应。
C) 氢键则进一步稳定了螺旋构象。圆二色谱实验发现,随着链长逐渐增加,Cotton效应不断增强,说明侧链的手性会诱导螺旋管产生手性,并且具有协同效应。
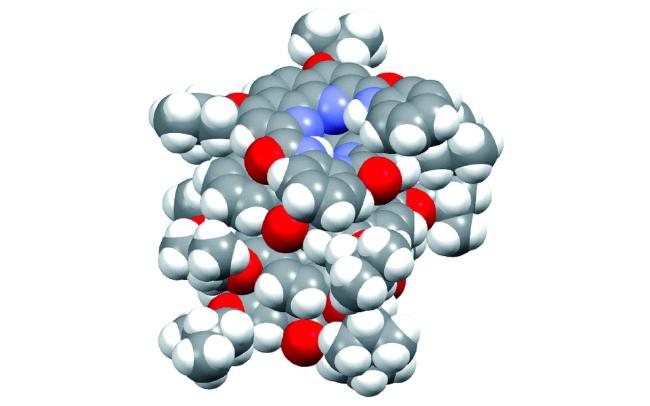
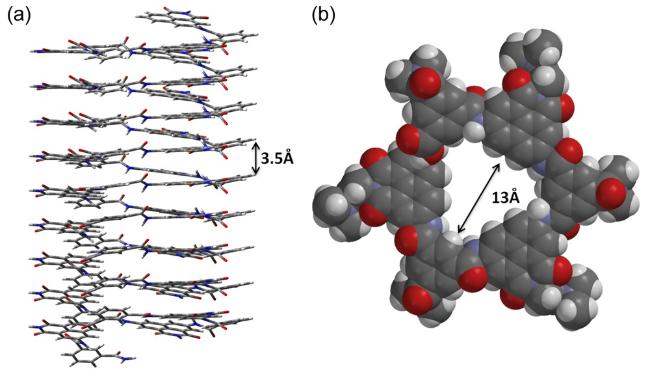
聚合物P28结构中设计的分子内氢键是形成螺旋结构的关键因素,其固定了聚酰胺的构象。没有分子内氢键存在下,酰胺寡聚体在氯仿中倾向于通过酰胺键的分子间N—H…O( C)氢键作用发生二聚形成同源双链[70]。对两亲性聚合物P29的研究进一步展现了不存在分子内氢键的聚芳酰胺在溶液中的折叠行为[71]。聚合物P29a的数均分子量Mn和重均分子量Mw分别为32.0和59.0 kDa(PDI=1.8)。聚合物P29a在水溶液及DMF、MeOH、CHCl3、CH2Cl2等多种不同极性的有机溶剂中均形成了螺旋结构(图4)。对聚合物P29a进行结构模拟可见,引入的较大尺寸芳香萘二胺单元不仅提供了更好的层间芳香堆积,还调控骨架折叠形成了1.3 nm的较大内径,螺旋结构的螺距为3.5 Å,其形成了9圈螺旋,因此螺旋管的深度达到3.1 nm。聚合物P29a的酰胺氮原子上甲基化得到P29b。P29a和P29b在不同极性溶剂中表现不同折叠行为,在低极性的二氯甲烷、氯仿等有机溶剂中,甲基化的P29b形成螺旋构象的倾向大大降低,说明除了疏溶剂作用,螺旋层间的分子内N—H…O(
C)氢键作用发生二聚形成同源双链[70]。对两亲性聚合物P29的研究进一步展现了不存在分子内氢键的聚芳酰胺在溶液中的折叠行为[71]。聚合物P29a的数均分子量Mn和重均分子量Mw分别为32.0和59.0 kDa(PDI=1.8)。聚合物P29a在水溶液及DMF、MeOH、CHCl3、CH2Cl2等多种不同极性的有机溶剂中均形成了螺旋结构(图4)。对聚合物P29a进行结构模拟可见,引入的较大尺寸芳香萘二胺单元不仅提供了更好的层间芳香堆积,还调控骨架折叠形成了1.3 nm的较大内径,螺旋结构的螺距为3.5 Å,其形成了9圈螺旋,因此螺旋管的深度达到3.1 nm。聚合物P29a的酰胺氮原子上甲基化得到P29b。P29a和P29b在不同极性溶剂中表现不同折叠行为,在低极性的二氯甲烷、氯仿等有机溶剂中,甲基化的P29b形成螺旋构象的倾向大大降低,说明除了疏溶剂作用,螺旋层间的分子内N—H…O( C)氢键是稳定螺旋构象的决定因素。在甲醇等大极性溶剂中,疏溶剂作用是形成螺旋结构的决定因素,这时聚合物P29a和P29b有类似的折叠行为。萘二胺单元的1,8-酰亚胺基团对聚合物形成螺旋结构也起到重要作用,没有1,8-酰亚胺基团时,聚合物不能自行折叠形成螺旋结构。在氯仿中聚合物与l-天冬氨酸等手性氨基酸二价盐通过多价分子间氢键诱导产生手性螺旋结构[72]。70% ee值的天冬氨酸负离子就能诱导聚合物产生最强Cotton效应,说明在螺旋形成过程中遵循多数决原则。螺旋的手性还与溶剂有很大关系,在氯仿中逐渐加入乙腈将改变螺旋结构的手性,在乙腈溶液中产生与氯仿溶剂中完全相反的手性。由间-苯二胺与2,2'-联吡啶单元交替偶联形成的聚芳酰胺在水溶液中也能形成螺旋结构[73]。苯基侧链的缬氨酸单元将手性传递给螺旋结构。由于联吡啶与Ni2+配位而改变构象,从而产生了手性相反的2种螺旋结构。
C)氢键是稳定螺旋构象的决定因素。在甲醇等大极性溶剂中,疏溶剂作用是形成螺旋结构的决定因素,这时聚合物P29a和P29b有类似的折叠行为。萘二胺单元的1,8-酰亚胺基团对聚合物形成螺旋结构也起到重要作用,没有1,8-酰亚胺基团时,聚合物不能自行折叠形成螺旋结构。在氯仿中聚合物与l-天冬氨酸等手性氨基酸二价盐通过多价分子间氢键诱导产生手性螺旋结构[72]。70% ee值的天冬氨酸负离子就能诱导聚合物产生最强Cotton效应,说明在螺旋形成过程中遵循多数决原则。螺旋的手性还与溶剂有很大关系,在氯仿中逐渐加入乙腈将改变螺旋结构的手性,在乙腈溶液中产生与氯仿溶剂中完全相反的手性。由间-苯二胺与2,2'-联吡啶单元交替偶联形成的聚芳酰胺在水溶液中也能形成螺旋结构[73]。苯基侧链的缬氨酸单元将手性传递给螺旋结构。由于联吡啶与Ni2+配位而改变构象,从而产生了手性相反的2种螺旋结构。
C)氢键作用发生二聚形成同源双链[70]。对两亲性聚合物P29的研究进一步展现了不存在分子内氢键的聚芳酰胺在溶液中的折叠行为[71]。聚合物P29a的数均分子量Mn和重均分子量Mw分别为32.0和59.0 kDa(PDI=1.8)。聚合物P29a在水溶液及DMF、MeOH、CHCl3、CH2Cl2等多种不同极性的有机溶剂中均形成了螺旋结构(图4)。对聚合物P29a进行结构模拟可见,引入的较大尺寸芳香萘二胺单元不仅提供了更好的层间芳香堆积,还调控骨架折叠形成了1.3 nm的较大内径,螺旋结构的螺距为3.5 Å,其形成了9圈螺旋,因此螺旋管的深度达到3.1 nm。聚合物P29a的酰胺氮原子上甲基化得到P29b。P29a和P29b在不同极性溶剂中表现不同折叠行为,在低极性的二氯甲烷、氯仿等有机溶剂中,甲基化的P29b形成螺旋构象的倾向大大降低,说明除了疏溶剂作用,螺旋层间的分子内N—H…O( C)氢键是稳定螺旋构象的决定因素。在甲醇等大极性溶剂中,疏溶剂作用是形成螺旋结构的决定因素,这时聚合物P29a和P29b有类似的折叠行为。萘二胺单元的1,8-酰亚胺基团对聚合物形成螺旋结构也起到重要作用,没有1,8-酰亚胺基团时,聚合物不能自行折叠形成螺旋结构。在氯仿中聚合物与l-天冬氨酸等手性氨基酸二价盐通过多价分子间氢键诱导产生手性螺旋结构[72]。70% ee值的天冬氨酸负离子就能诱导聚合物产生最强Cotton效应,说明在螺旋形成过程中遵循多数决原则。螺旋的手性还与溶剂有很大关系,在氯仿中逐渐加入乙腈将改变螺旋结构的手性,在乙腈溶液中产生与氯仿溶剂中完全相反的手性。由间-苯二胺与2,2'-联吡啶单元交替偶联形成的聚芳酰胺在水溶液中也能形成螺旋结构[73]。苯基侧链的缬氨酸单元将手性传递给螺旋结构。由于联吡啶与Ni2+配位而改变构象,从而产生了手性相反的2种螺旋结构。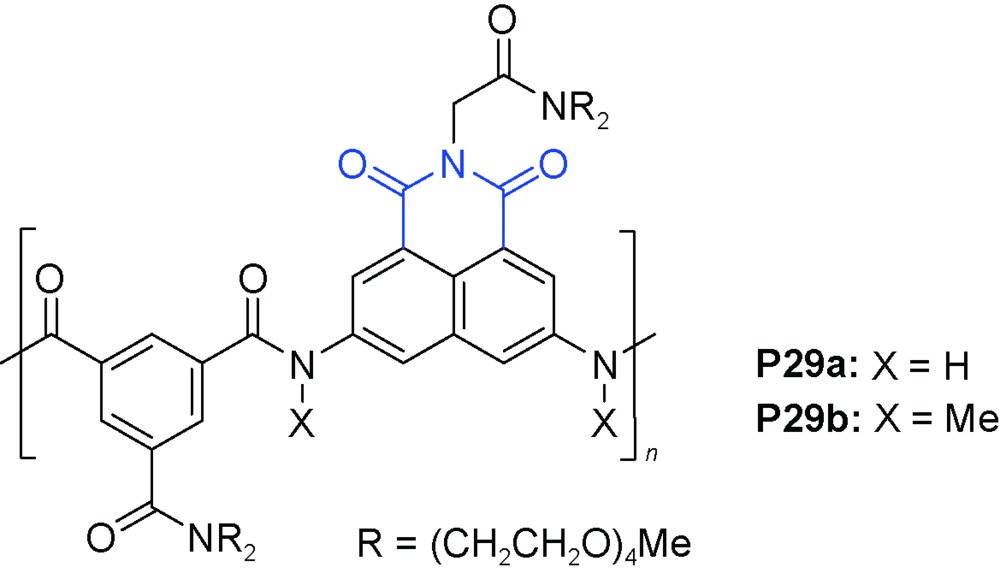
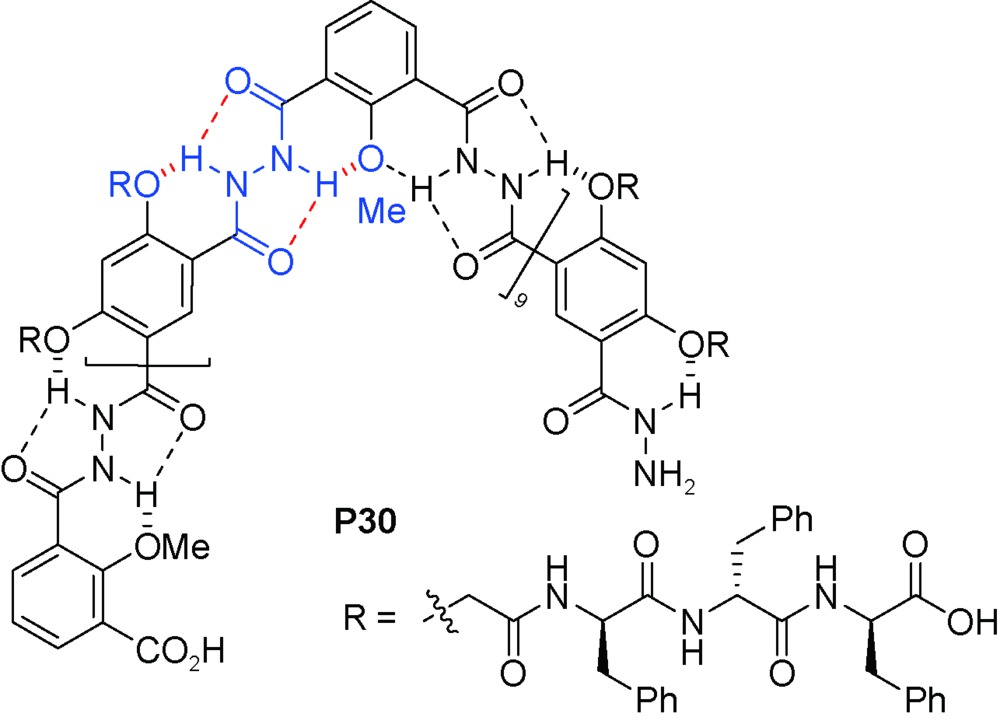
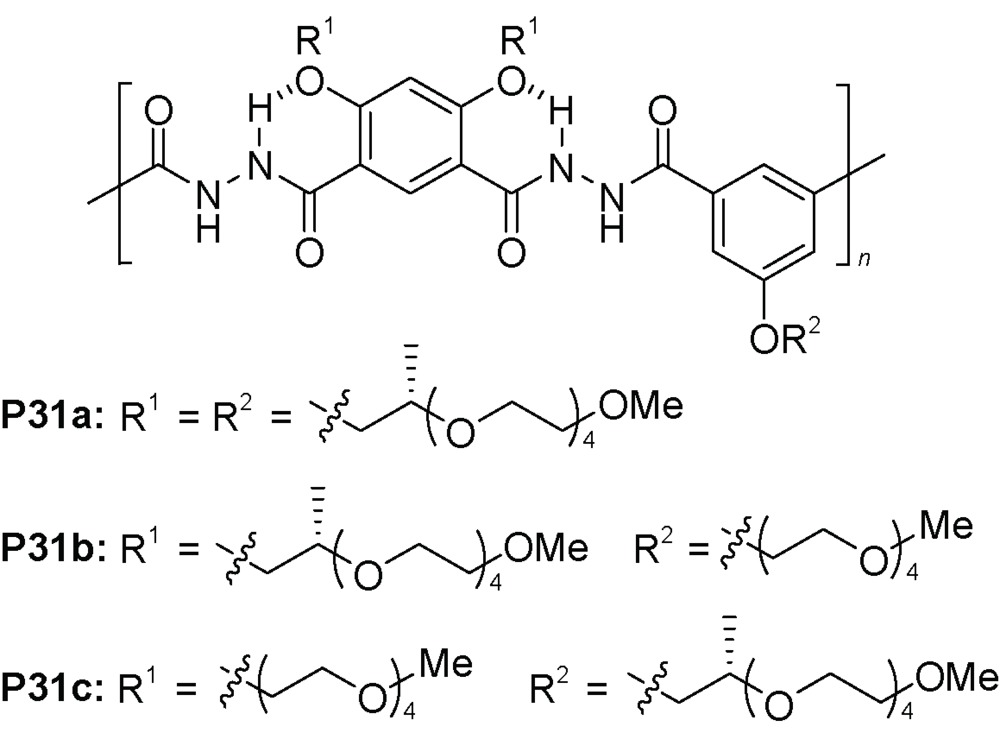
芳酰肼聚合物P31a~c的数均分子量Mn分别为14 365、12 209和7201,重均分子量Mw分别达到31 200、31 087和21 495(PDI分别为2.17、2.55、2.99)[76]。部分重复单元的分子内氢键使聚合物有一定刚性,引入的多个缩乙二醇侧链使其溶于水及EtOH、MeOH、CHCl3、CH2Cl2等多种有机溶剂。由于疏水作用,聚合物P31a和P31b形成有约4圈螺旋的螺旋管结构,较短的P31c形成有约2圈螺旋。聚合物P31a和P31b的手性侧链也使得螺旋管具有单一手性倾向,圆二色谱中表现出相当的Cotton效应。说明聚合物的聚合度及手性侧链所处的位置对于手性传递至关重要,手性螺旋构象的形成与具有分子内氢键的刚性结构单元相关联,而没有分子内氢键的柔性结构单元的贡献较小。
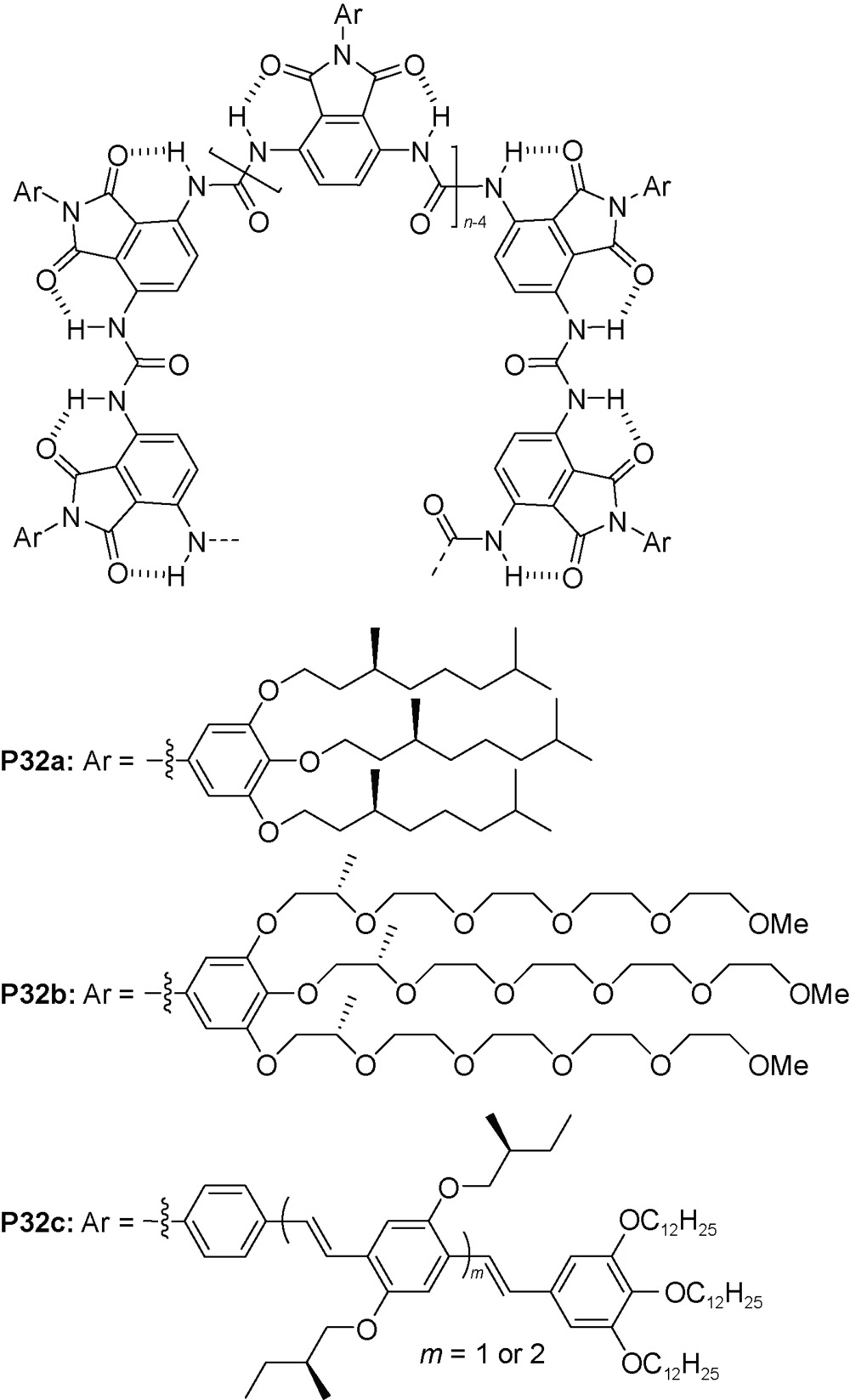
3,6-二氨基-1,2-苯二甲酰亚胺和相应的二异氰酸单体聚合得到聚合物P32a~c,分子内N—H…O( C) 氢键使聚合物有刚性结构。聚合度较高的聚合物P32a(n=~30) 在THF中形成手性螺旋[77]。尿素单元形成顺式构象,每6~8个重复单元形成一圈螺旋,所以聚合物P32a约形成4~5圈螺旋,层间芳香堆积作用是螺旋构象的重要稳定因素。而在CHCl3溶剂中,由于分子内N—H…O(
C) 氢键使聚合物有刚性结构。聚合度较高的聚合物P32a(n=~30) 在THF中形成手性螺旋[77]。尿素单元形成顺式构象,每6~8个重复单元形成一圈螺旋,所以聚合物P32a约形成4~5圈螺旋,层间芳香堆积作用是螺旋构象的重要稳定因素。而在CHCl3溶剂中,由于分子内N—H…O( C) 氢键被破坏,聚合物P32a不能形成螺旋结构。
C) 氢键被破坏,聚合物P32a不能形成螺旋结构。
C) 氢键使聚合物有刚性结构。聚合度较高的聚合物P32a(n=~30) 在THF中形成手性螺旋[77]。尿素单元形成顺式构象,每6~8个重复单元形成一圈螺旋,所以聚合物P32a约形成4~5圈螺旋,层间芳香堆积作用是螺旋构象的重要稳定因素。而在CHCl3溶剂中,由于分子内N—H…O( C) 氢键被破坏,聚合物P32a不能形成螺旋结构。引入亲水性侧链使聚合物P32b(n=~20) 在水溶液中有一定溶解度,圆二色谱显示,其有两种Cotton效应信号,并且不随测试温度的变化而变化,疏水作用是形成螺旋结构的驱动力[78]。而在THF溶剂中,聚合物P32b只有一个Cotton效应信号,且符号与水溶液中相反,说明溶剂对螺旋手性起到决定作用。当测试温度逐渐升高时,Cotton效应逐渐减弱;当温度升高至55 ℃时,Cotton效应完全消失。这说明聚合物构象在THF中存在动力学平衡,随着温度升高,聚合物构象可能逐渐转化为无规卷曲,或者形成了等量的M和P两种手性螺旋。
引入对苯撑乙烯寡聚体(OPV) 侧链的聚合物P32c能够溶于多数有机溶剂[79]。与P32a相类似,P32c在THF中形成手性螺旋,而在CHCl3溶剂中以无规卷曲结构存在。有趣的是,当不同溶剂中的样品分别浓缩后再溶解于正庚烷,在之前所溶解的溶剂中的构象将保持到正庚烷溶剂中。紫外-可见光谱实验表明具有大共轭体系的OPV侧链之间有芳香堆积,然而聚合物P32c的Cotton效应比较弱,说明这些侧链之间可能存在无序的集聚。
3.2 三氮唑和口恶二唑构筑基元
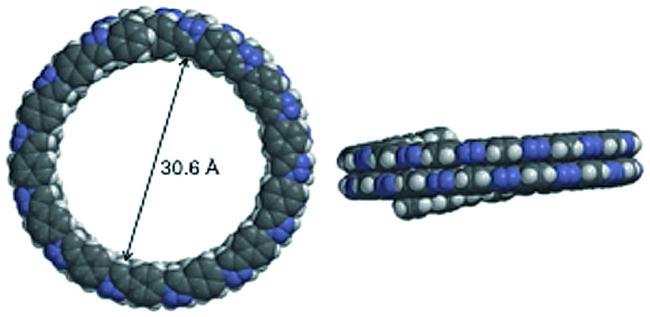
与寡聚体9的结构相类似,苯和三唑/吡啶重复单元构建的聚合物P33a和P33b(Mw=(1.6~5.3)×103 g/mol, PDI=1.5~2.3) 在CH2Cl2或CH3CN等溶剂中也形成螺旋结构[80]。聚合物的苯和吡啶单元都采取间位取代形式,三唑/吡啶采取反式构象[81]。在疏溶剂作用驱动下,π-π堆积稳定了螺旋构象,手性侧链使螺旋具有手性。对位取代的苯基与1,4-二取代-1,2,3-三唑交替聚合得到聚合物P34a,b和P35[82]。当苯基的两个三唑取代基采取顺式构象时,聚合物链弯曲形成螺旋结构,其弯曲的弧度要小于间位取代的聚合物P33a,b。由于弧度小,每圈螺旋约有14.5个重复单元,螺旋的内径有相当大的尺寸,可以达到30.6 Å(图6)。促使采取这种顺式构象的重要因素是溶剂。在DMF中,聚合物倾向于形成无规卷曲;当水的含量逐渐增加,聚合物倾向于形成紧密的螺旋结构,螺距变小,并自组装成更长的螺旋管结构。当水含量进一步增加时,这些螺旋管自组装成螺旋管束。螺旋的手性来源于手性侧链。聚(γ-苄基-l-谷氨酸)(PBLG)诱导了非手性侧链取代的聚合物P35产生手性螺旋。PBLG具有α-螺旋结构,直径为1.6 nm,由于疏溶剂作用与聚合物P35在含水量超过18%的DMF溶液中组装成套管结构,并诱导P35产生手性。
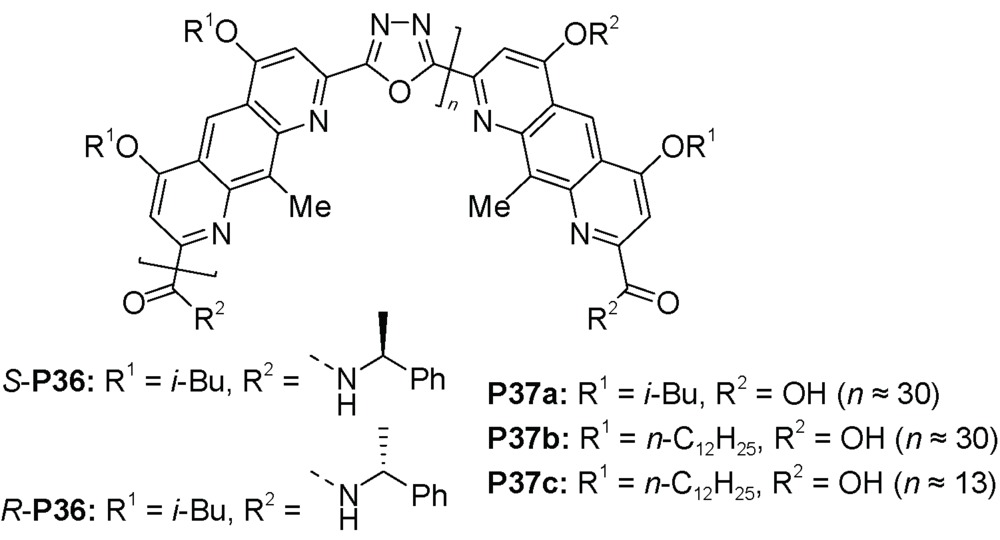
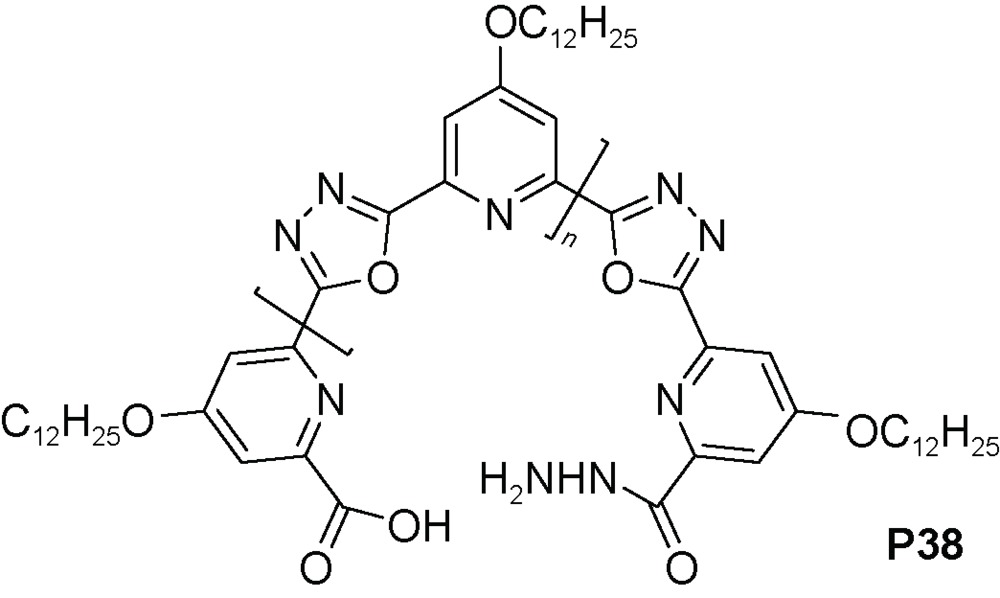
与寡聚体16和17类似,聚合物P36和P37也形成螺旋结构,在CH2Cl2中S-P36和R-P36有互为镜像的Cotton效应。在CH3CN/H2O两元溶剂中,随着水的含量增加,P37a自组装为双螺旋结构。P37a随浓度升高也倾向于形成双螺旋。而随着温度升高,双螺旋又会转化为单股螺旋。隧道扫描显微镜实验展现了单分子水平的单股螺旋和双螺旋结构。聚合物P37b和P37c引入了长的脂肪链侧链,保证了其脂溶性[83]。较长的聚合物P37b形成了内径约为4.8 Å、长约3.3 nm的螺旋管,长度与脂双层的厚度相当(3.5 nm),恰好能够形成单分子跨膜通道。P37c螺旋管长约1.4 nm,其跨膜通道由分子间π-π堆积组装而成。相对来说,P37b更稳定,表现出与天然通道分子相比拟的寿命和传输效率。部分酸化的2,6-吡啶二酸钠盐与2,6-吡啶二酰肼在偶联试剂DEPBT作用下,能够以64%收率得到Mn达到9.2 kDa的聚吡啶酰肼,重复单元达27个。在POCl3作用下脱水,一锅煮高效制备得到聚(吡啶/1,3,4-口恶二唑)P38[85]。改变单体及缩合剂能够得到不同长度的聚合物,这成为一种高效的长螺旋管构建策略。P38有亲水性富电子的空腔,氮原子和氧原子交替排列构象为最稳定构象,直径约3 Å,平均长度2.7 nm,作为离子通道对K+/Na+表现出优秀的K+选择性。
3.3 间-苯乙炔和杂环类似物构筑基元
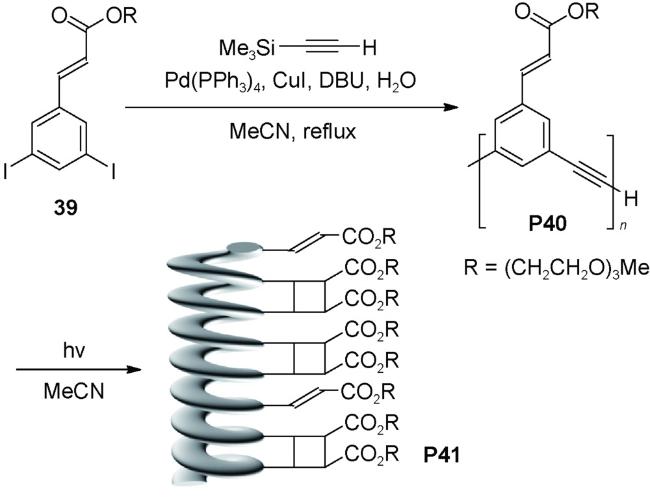
最早构筑聚合物螺旋管的实例来自间-苯乙炔类聚合物[86]。如图式2所示,间位二碘代的肉桂酸酯39,与乙炔基三甲基硅烷在Pd(PPh3)4/CuI作用下偶联聚合,得到聚合度达到60的聚合物P40(PDI=1.3)。在乙腈中,P40因疏溶剂作用折叠形成螺旋结构,每一圈螺旋有6个重复单元,螺距约3.4 Å,螺旋管的长度达到3.4 nm。由于协同作用,螺旋结构的稳定性优于相应苯乙炔寡聚体。短时间光照下,处于螺旋外围的乙烯基侧链发生分子内[2+2]环加成反应得聚合物P41,因侧链的共价固定,螺旋结构更加稳固,加入CHCl3不会破坏螺旋结构。
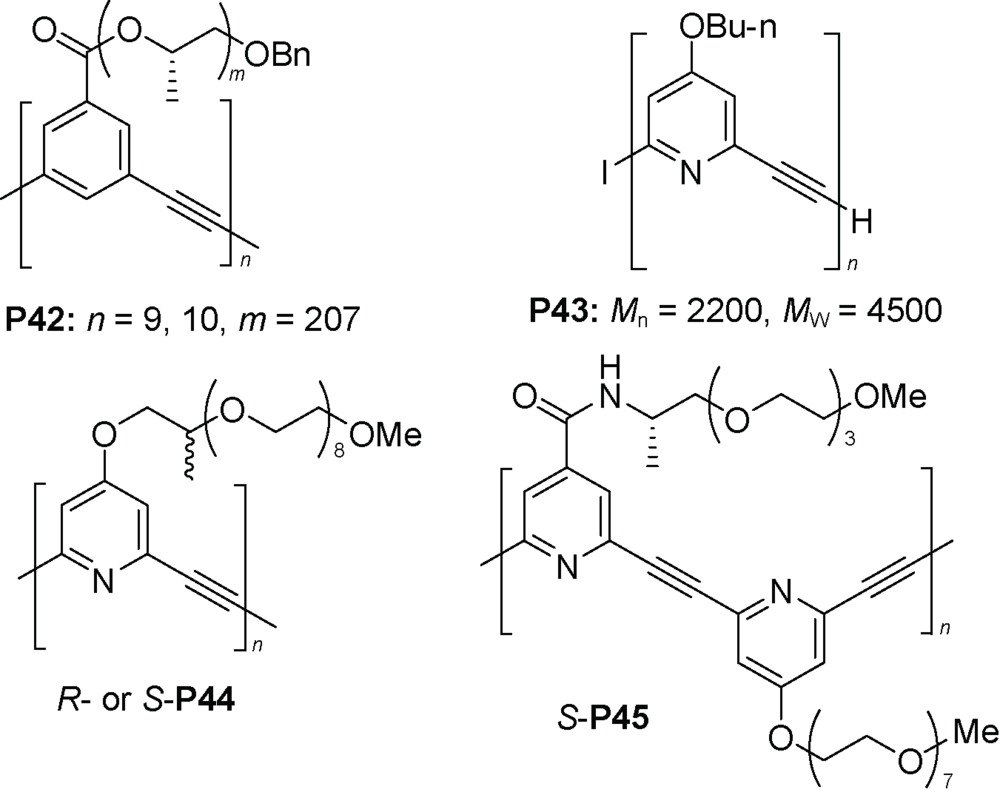
等规立构的聚环氧丙烷(PPO)和聚间-苯乙炔(PmPE)的两亲性接枝共聚物P42,芳香骨架的聚合度较低[87]。在CHCl3中,P42的光学活性较弱,说明其倾向于采取伸展的构象以避免PPO侧链之间的位阻效应;在极性的MeOH或CH3CN溶剂中,光学活性有所增强。然而这种信号并非来源于PmPE,而是来源于PPO片段。PmPE要形成螺旋结构,层间的芳香堆积是重要因素,这种堆积要求芳环之间相距约3.4 Å,但是PPO堆积的最小距离是4.7 Å,阻碍了芳香片段的折叠。聚乙炔基吡啶P43在CH2Cl2中也具有高度流动性构象[61]。与乙炔基吡啶寡聚体20a和20b类似,P43作为主体分子,受吡喃糖苷诱导在CH2Cl2等低极性溶剂中形成手性螺旋结构。并且,P43能够将水溶性天然单糖分子萃取到氯仿等低极性有机溶剂。在吡啶环上引入水溶性缩乙二醇侧链,相应两亲性聚合物能够在质子性溶剂中形成螺旋结构[88]。这一螺旋结构的驱动力主要来自芳香堆积作用。在MeOH/H2O(10∶1) 中,其与天然单糖形成络合物,单糖的手性传递给非手性聚合物使之产生手性螺旋管。在质子性溶剂中,这种络合源自单糖与聚合物中吡啶单元的氮原子之间的氢键作用。在吡啶环上引入手性缩乙二醇侧链,得到水溶性的手性聚合物R-和S-P44[89]。不同聚合度的组分、不同测试温度下有不同的CD信号,说明它们表现出不同构象。高分子量组分聚合物可达67-聚体,其自身可能形成单分子双螺旋或更为复杂的构象形式。高分子量组分在水溶液中与甘露糖、环糊精或糖原等多糖结合,其诱导的CD信号与高温条件下或者低分子量组分的CD信号相类似,说明在这些条件下都形成单股螺旋结构。聚合物S-P45有一条手性的酰胺缩乙二醇取代基和一条非手性的缩乙二醇取代基,使之溶于极性和非极性溶剂中[90]。这个手性聚合物在CD谱中信号比较弱,酰胺键的引入使聚合物可以在水或乙醇水溶液中与金属盐,特别是Sc(OTf)3,配位而稳定螺旋结构。与等当量的金属离子配位后,聚合物在CD谱中有最强的正Cotton效应。添加了Sc(OTf)3后紫外可见光谱中观察到360 nm处的减色效应,说明Sc3+与酰胺键发生了配位作用。Sc(OTf)3与侧链的酰胺键发生配位,有助于π-π堆积的形成,从而稳定了螺旋结构。
4 芳香超分子螺旋管
无论是寡聚体还是聚合物,要得到具有可利用的内径和足够长度的螺旋管并不容易,超分子策略可能成为一种有效方法。环肽线性堆积是构建分子管结构的一种方案[7],然而在生理环境条件下,多种弱相互作用力的协同作用对构筑超分子螺旋管带来挑战。
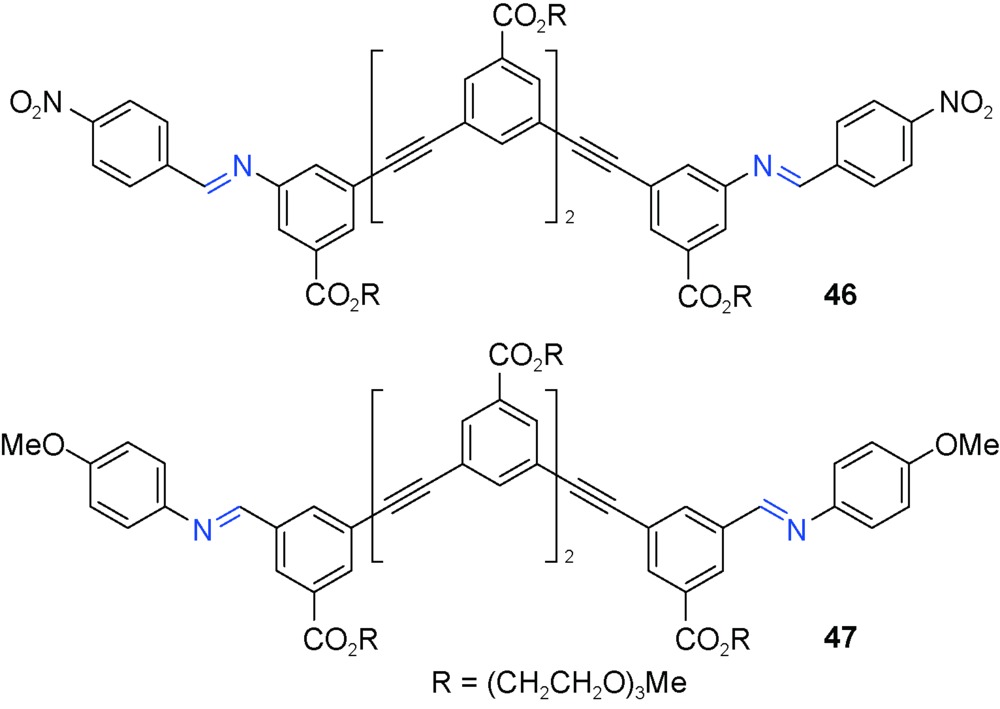
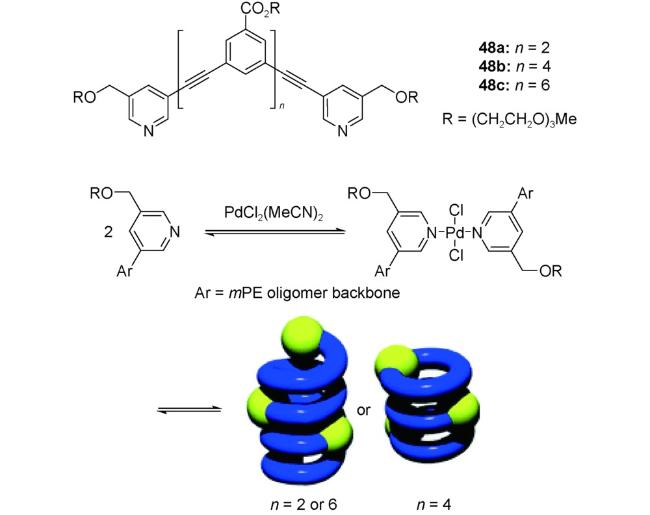
配位作用是超分子折叠体自组装的一种重要作用力,多样的几何构型能够诱导低聚物进行有序折叠[15]。吡啶的氮原子不仅可以作为氢键受体,还是良好的金属配位位点。将苯乙炔寡聚体2个端位的苯基置换为吡啶制得的寡聚体48a~c,由于链长较短,自身不具有稳定优势构象[93]。当寡聚体的乙腈溶液中加入反式-PdCl2(CH3CN)2,由于Pd2+与寡聚体的吡啶氮原子发生配位而自组装。如图式3所示,对于四聚体和八聚体,其通过首尾连接组装成超分子聚合物螺旋管,层间发生π-π堆积。而对于六聚体,则首先发生Pd2+与48b自身首尾吡啶氮原子的配位形成环状结构,环状配位化合物由于疏溶剂作用再发生线性堆积得到柱状管结构。亚胺键或者金属配位键的引入并未改变所得到的超分子螺旋管的本质,因为它们仍然可以作为一些线性客体或小分子的主体化合物。
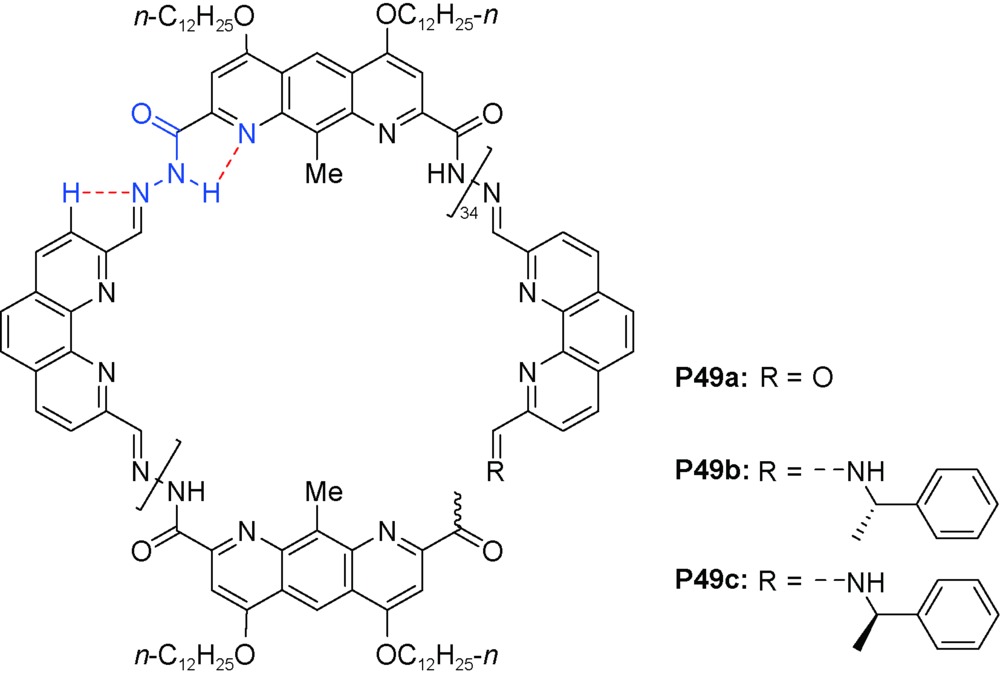
动态共价超分子聚合物P49a由2,8-吡啶并[3,2-g]喹啉二酰肼和2,9-二氮杂菲二醛缩合而成,Mn达到31 kDa(PDI=1.46)[94]。由于电子效应、位阻效应及分子内氢键作用等多种作用力,使酰腙单元采取反式构象,P49a倾向于形成螺旋二级结构。端位引入手性基元的聚合物P49b和P49c的CH2Cl2溶液在圆二色谱中表现出互成镜像的Cotton效应。这一酰腙螺旋管的内径达到1.0 nm,螺旋管的空腔结构类似巯基过氧化酶,这为催化反应提供了一个类酶微环境。以CHCl3/正己烷(3∶1) 为溶剂,P49a成功地催化了H2O2氧化苯硫酚PhSH生成氧化偶联产物PhSSPh,而其单体则不能催化这一氧化过程。在低极性有机溶剂中,P49a的有序二级结构为大极性的反应物提供了反应场所。
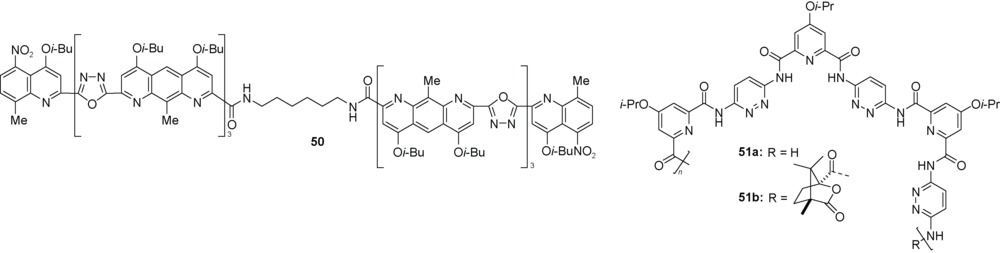
除了形成动态共价键或者金属配位键以外,其他在超分子构建中常见的弱相互作用力虽然强度有限,但是经过精巧设计仍能够成为构筑超分子聚合物的重要推动力。寡聚体50由2条吡啶并[3,2-g]喹啉/1,3,4-口恶二唑寡聚体与己二胺缩合而成[95]。寡聚体片段折叠形成螺旋结构,由于溶剂、浓度、温度等因素能够促使这种单股螺旋通过π-π堆积二聚为双螺旋[84],通过NMR、MALDI-TOF MS、AFM、TEM、SEM、DLS等实验证实,寡聚体50通过π-π堆积形成了线型超分子聚合物。哒嗪/吡啶/哒嗪砌块也具有弯月型构象[96]。2,6-吡啶二酸和3,6-哒嗪二胺缩合得到的寡聚体51a,其聚合度约为15,Mn为3000(PDI=1.17),理论计算表明其形成三角形的螺旋结构,内径约为0.6 nm,每圈螺旋有3.5个重复单元,因此分子内π-π堆积作用也是稳定螺旋构象的重要因素。在CH2Cl2中,端位引入樟脑基的手性寡聚体51b有强Cotton效应。寡聚体51a的螺旋构象使芳杂环的氮原子都朝向螺旋管内部,从而能够识别碱金属离子,由于单个螺旋管的长度约为1.5 nm,其作为跨膜离子通道时通过分子间π-π堆积形成了超分子螺旋管结构。
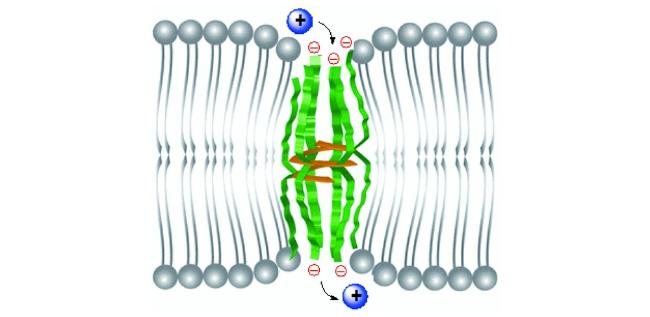
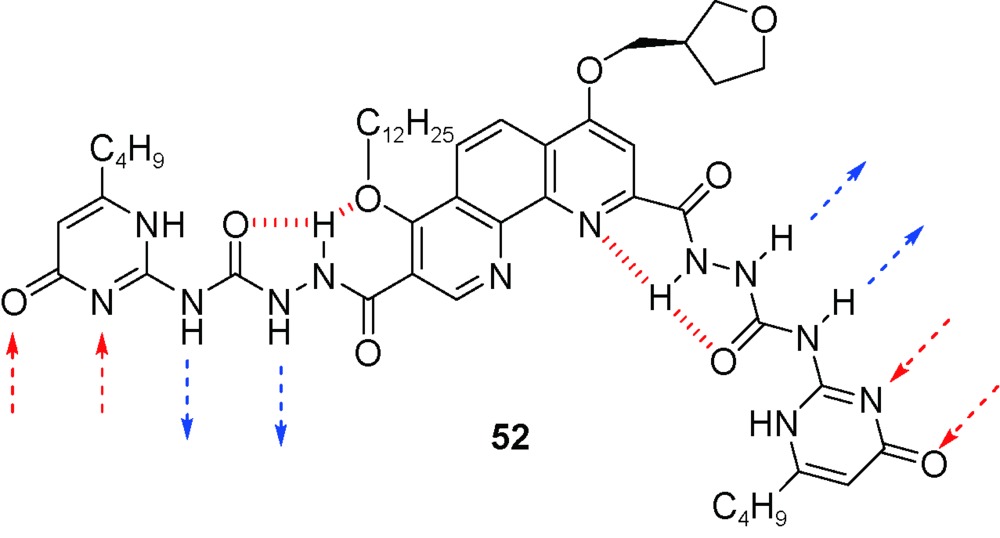
脲基嘧啶酮(UPy) 衍生物的分子间AADD·DDAA四重氢键作用是设计超分子化合物的一种有效手段。将这一基元引入邻二氮杂菲骨架,利用分子内氢键固定分子形状(52),从而构建了一类分子间多重氢键驱动的自组装超分子纳米管结构[97]。52自组装而成的超分子聚合物Mn达到2.37×105(PDI=1.17),内径约2.6 nm,螺旋管长度达到8.2 nm。这一砌块成功地在脂双层自组装形成分子通道,较大内径使其具备转运葡萄糖分子的功能,这也提供了溶液相氢键自组装形成螺旋管的策略。
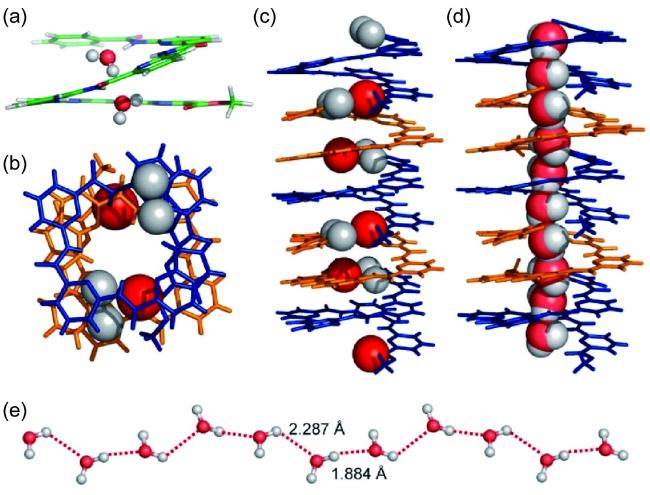
水通道蛋白在水的跨膜转运过程中起关键作用。吡啶五聚体7的螺旋结构空腔中结合水分子,五聚体7线性排列将可能模拟水通道蛋白的功能。设计寡聚体之间发生分子间非共价作用力能够实现这一排列形式[17]。五聚体53a和53b的设计实现了分子间“端到端”的粘结[98]。不同于吡啶寡聚体7,在53a的C-端引入甲氧酰基,羰基氧原子C  O与另一个53a分子N-端苄氧羰基的苯基CH形成分子间(Bn)C—H…O(
O与另一个53a分子N-端苄氧羰基的苯基CH形成分子间(Bn)C—H…O(  C)氢键作用,这种弱相互作用(晶体结构显示,氢键键长为2.44 Å)使53a分子之间发生“端到端”的粘结,所有寡聚体片段均有相同的手性,从而构建了超分子螺旋管。53a的超分子螺旋体空腔中形成了线性排列的甲醇分子(图7)。在含水溶剂中进行单晶培养,空腔中主要结合的仍是甲醇分子,仅有24%~40%的甲醇被置换为水分子。说明对小分子的包结有高度选择性。将N-端的苄氧羰基置换为苯酰基的53b,其通过分子间三中心(Ph)H2…O(
C)氢键作用,这种弱相互作用(晶体结构显示,氢键键长为2.44 Å)使53a分子之间发生“端到端”的粘结,所有寡聚体片段均有相同的手性,从而构建了超分子螺旋管。53a的超分子螺旋体空腔中形成了线性排列的甲醇分子(图7)。在含水溶剂中进行单晶培养,空腔中主要结合的仍是甲醇分子,仅有24%~40%的甲醇被置换为水分子。说明对小分子的包结有高度选择性。将N-端的苄氧羰基置换为苯酰基的53b,其通过分子间三中心(Ph)H2…O(  C) 氢键的粘结形成线性结构(晶体结构显示,氢键键长为2.75 Å和2.82 Å),其空腔内径仅为2.8 Å,只能容纳水分子而形成水线(图8)[99]。相应的四聚体或者六聚体都不能发生这样的粘结,“端到端”发生粘结的两个氢键给受体也需要在电子效应和位阻效应方面相匹配。
C) 氢键的粘结形成线性结构(晶体结构显示,氢键键长为2.75 Å和2.82 Å),其空腔内径仅为2.8 Å,只能容纳水分子而形成水线(图8)[99]。相应的四聚体或者六聚体都不能发生这样的粘结,“端到端”发生粘结的两个氢键给受体也需要在电子效应和位阻效应方面相匹配。
O与另一个53a分子N-端苄氧羰基的苯基CH形成分子间(Bn)C—H…O( C)氢键作用,这种弱相互作用(晶体结构显示,氢键键长为2.44 Å)使53a分子之间发生“端到端”的粘结,所有寡聚体片段均有相同的手性,从而构建了超分子螺旋管。53a的超分子螺旋体空腔中形成了线性排列的甲醇分子(图7)。在含水溶剂中进行单晶培养,空腔中主要结合的仍是甲醇分子,仅有24%~40%的甲醇被置换为水分子。说明对小分子的包结有高度选择性。将N-端的苄氧羰基置换为苯酰基的53b,其通过分子间三中心(Ph)H2…O( C) 氢键的粘结形成线性结构(晶体结构显示,氢键键长为2.75 Å和2.82 Å),其空腔内径仅为2.8 Å,只能容纳水分子而形成水线(图8)[99]。相应的四聚体或者六聚体都不能发生这样的粘结,“端到端”发生粘结的两个氢键给受体也需要在电子效应和位阻效应方面相匹配。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
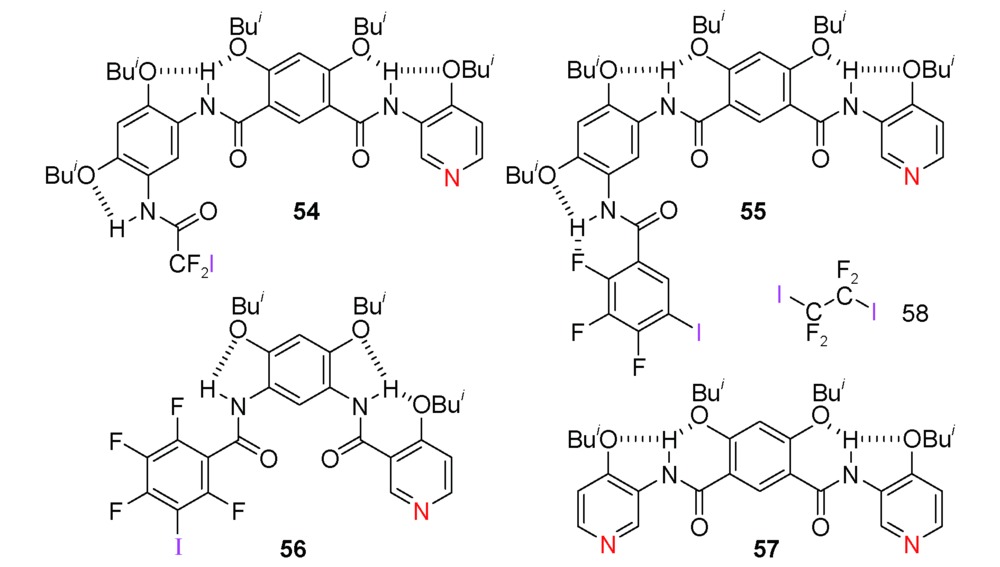
卤键作为一种与氢键类似的弱相互作用,也成功用于超分子螺旋体的构建[100]。在芳酰胺寡聚体54~57结构中,分子内氢键使其具有弯月型构象,在寡聚体的端基分别引入卤键给体和(或)卤键受体,可能存在多种结合形式。分子间依次发生首尾卤键作用,可能形成线性卤键聚合物;也可能通过[1+1]等分子间相互作用形成大环分子。寡聚体54~56的单晶结构表明,两分子寡聚体通过分子间C—I…N卤键的作用首尾相连,且由于单体的弯月型构象而形成螺旋体,并进一步组装成双股螺旋和四股螺旋。寡聚体57的两端均是卤键受体,其与58在固相中形成2+2模式的依次作用,交替排列形成四股螺旋。这类超分子螺旋体的构筑中,分子内氢键和分子间卤键表现出正交性质,芳香堆积作用协同参与,形成了高度有序的高级结构。通过单体的设计可能获得具有可利用空腔的可溶性分子,并进行功能探索。进一步研究表明,利用卤键还可以控制超分子大环和螺旋管形成的选择性[101]。
5 结论与展望
芳香砌块是构筑超分子骨架的重要基元。线性芳香大分子在π-π堆积作用、氢键作用、静电斥力或配位作用等非共价作用驱动下,构建了多种类型的螺旋结构,足够长的螺旋体形成管状分子。分子管可以由较长单一分子或聚合度较大的聚合物构建,也可以由多个短的螺旋管自组装而成。动态共价键、配位作用、分子间π-π堆积、氢键和卤键作用等被证实为有效的超分子螺旋管构筑策略。芳香砌块的多样性及多种相互作用的选择可以设计并合成到尺寸可调并具有丰富功能的螺旋管结构。然而就目前的螺旋管的功能而言,主要还是集中在金属离子、卤素负离子、溶剂分子及单糖、双糖、多糖等客体分子的识别与包结;手性小分子或手性侧链的手性诱导等方面。虽然螺旋管模拟生物跨膜通道以及催化化学转化等方面也有若干报道,要实现更广泛的功能开发,螺旋管的设计和合成仍具有相当的挑战性。对螺旋管尺寸的精准设计、空腔内部官能团的定向引入等都将发展出螺旋管的新功能。目前要获得较长的螺旋管结构,在制备方面还没有实质性的突破。单体聚合时往往由于寡聚体的折叠构象引起位阻效应等阻碍了更大分子量聚合物的生成[102]。折叠构象在一定阶段有利于聚合物的生成,而发展为更大分子量的聚合物时又成为阻碍因素之一[103]。通过分子间弱相互作用将短的折叠体组装为长的螺旋管方面虽然已经取得了不少成果,但如何实现溶液相,特别是水溶液中的有效控制,还没有完全有效的方案,这些都有待于在理论和实践方面有所创新。