



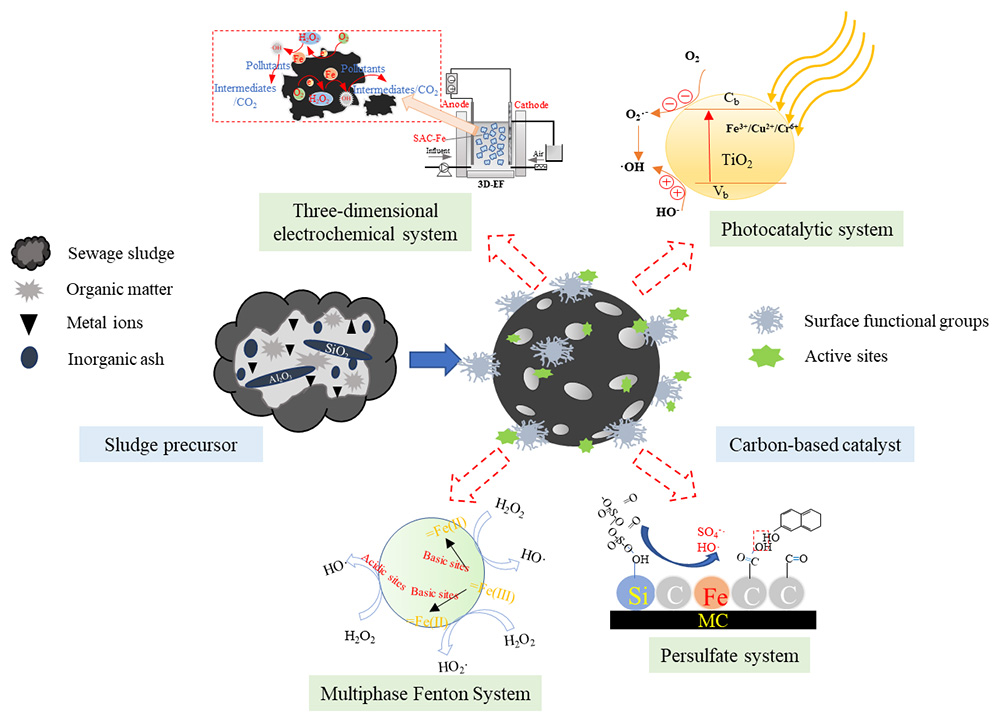
1 引言
城市污泥(Civic Sludge),也叫排水污泥(Sewage Sludge),是指在水处理过程中所形成的以有机物为主要成分的泥状物质,如得不到有效处理,将会对环境造成二次污染[1],直接影响我国水污染控制条例的实施与成效。当前污泥的资源化利用主要包括污泥土地利用、污泥制陶粒、污泥制砖、污泥制生态水泥、污泥低温热解制取燃料和污泥碳化等[2]。由于污泥含水率较高(95%~99.5%),加重了后期污泥处理、处置负担,因此强化污泥脱水是污泥资源化利用的重要手段[3]。脱水预处理一般包括化学、物理和生物调理法等[4~6],这些方法一方面改善了污泥的理化性质,使细胞结合水易于分离;另一方面也使得污泥菌胶团发生破解,污泥颗粒形态和表面化学基团发生改变。因此,强化污泥脱水引起的污泥性质变化(如污泥含水率降低、重金属总量改变等)也使后期的污泥热解碳化技术得以应用并出现了脱水与碳化的耦合技术。研究表明污泥碳化材料作为吸附剂已经在去除水中染料、持久性有机污染物、重金属等面发挥着重要作用,而近年来对污泥碳化制备高附加值产物的研究不断表明,以污泥作为原料生产碳基催化剂并用于水环境催化是一种新型的污泥减量和资源化方式。因此,基于污泥碳基功能材料发展出的催化剂近期被广泛的应用于环境催化领域,尤其是针对持久性有机污染物[7]、带有共轭基团且毒性较强的苯系物和萘系物[8]以及硫化氢[9]、氮氧化物[10,11]等气态污染物等。
由于污泥组成性质复杂,以污泥制备的催化剂其活性组分的作用机制与催化过程及合成条件密切相关,尤其是涉及特异性吸附和相间电子转移。近年来,包括本课题组在内的许多学者都致力于寻找各种方法拓展污泥碳基材料在水环境催化领域的应用,如多相Fenton体系[8]、过硫酸盐体系[12,13]、光Fenton体系[14]、臭氧化体系[15]、湿式催化氧化体系[16]和电催化体系[17]等,同时利用各种研究手段探究污泥碳基材料形成过程与污泥中灰分、碳载体和活性组分化学形态之间的相关性,寻找对水中污染物降解效率的影响因素。因此,本文将在此基础上,详细阐述污泥碳基材料的制备方法与材料物理化学性质的构效关系,并结合其在不同催化体系中的应用和机理说明污泥碳基催化剂参与的吸附、电子转移和降解水中有机污染物的作用机制,同时对提高材料的利用效率、稳定性和可重复利用性提出新的展望。
2 污泥碳基前躯体的物理化学性质
污泥碳化产物形成催化剂前躯体的活性一般与其比表面积、孔隙率、物相组成以及元素分布、表面特征官能团和pH值有关,制备条件如高温热解温度、活化气氛、活化剂种类等均影响碳化产物的理化性质。典型的污泥碳基前躯体一般具有较高的比表面积、丰富的表面含氧官能团(羟基、羧基、酮基、内酯基等)和助催化剂(SiO2、Al2O3、CaO等)等成分。如Huang等[18]发现污泥碳材料表面的碱性官能团和离域π电子可作为活性位点催化过硫酸盐产生硫酸根自由基,而本课题组的研究结果表明灰分在污泥热转化时形成的磁性Fe3O4具有催化H2O2产生羟基自由基的特性[8]。近年来,许多研究都致力于改变制备方法以提高前躯体的比表面积,同时增强碳化产物的表面结构特性。
2.1 化学组成
与传统木质、煤、焦油等生物质制备的碳基材料不同,以剩余污泥为原料制备的生物碳H、O原子会与边缘碳原子以化学键的方式结合并形成丰富的表面官能团,如羧基、内酯基、羰基、酚羟基、吡啶、吡咯等。同时,污泥中各种灰分组成的水合无机氧化物在热解时则会通过脱水、缩合等形式形成新的矿物盐,成为活性位点沉积在碳晶表面或载体的孔道内。其中碳基载体中的主要无机物SiO2因具有石英的Si—O—Si键,遇水后表面羟基化,易与过氧化物形成氢键(如图1)加速中间体在载体上的吸附,促进中间体氧化物的形成;污泥载体中Al2O3因其pHIEP约为8.5~10.4,可提高污泥碳基催化剂的等电点,同时作为路易斯酸,吸引电子,降低Fe(Ⅲ)的稳定性,使Fe(Ⅲ)易于氧化H2O2并被还原为Fe(Ⅱ),提高污泥碳催化剂的催化效率[19]。污泥碳中的无机物包括Fe、Ca、Al、Si等含量还会影响污泥碳表面极性,对一些含羧基或酚酸基团的有机污染物分子产生特异性吸附;这些污泥无机灰分会与一些小分子的有机酸(草酸、乙酸等),如降解矿化产生的极性中间产物形成配位键,强化了催化时对后期COD的去除[4,20],增强污泥碳的吸附催化过程。
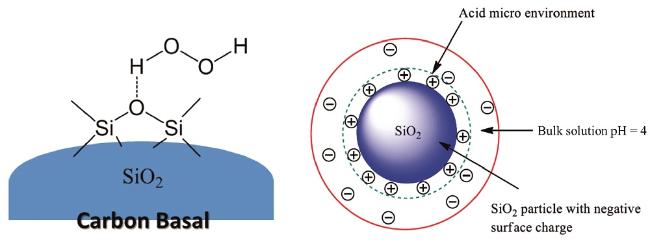
除污泥中常见的CaO、SiO2、Al2O3、Fe2O3等无机氧化物外,Ros等[21]还发现高温过程可以加速矿物熔融,形成结构更为复杂的化合物,其中,含量较高的钠长石(NaAlSi3O8)、方解石(CaCO3)等会沿碳基孔壁交叉生长,并在热解时催化碳基释放气体,降低载体中碳元素含量,并增加催化剂的孔体积。
2.2 孔隙结构
污泥碳化产物随制备工艺改变而拥有差异化的孔隙结构与微观形貌。碳化物的孔结构决定其吸附强弱,进一步影响其催化活性。活化试剂的种类和浓度、碳化温度、酸洗条件等均能改变碳基材料的比表面积,从而影响其作为催化剂对氧化剂和降解底物的吸附。
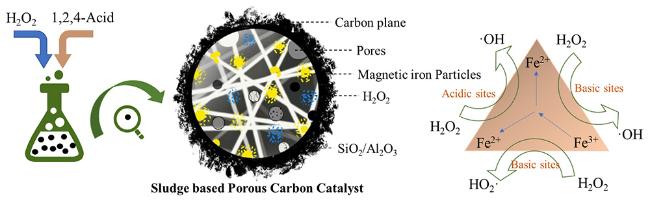
为获得高比表面积的污泥碳,研究者常使用KOH、ZnCl2、K2CO3、Fenton试剂等活化污泥,再控制一定的热解温度(400~800 ℃),辅以酸洗和二次活化[32]。200~400 ℃的低温段属于污泥中水分和小子分有机酸的挥发段,该温度区间碳载体上较易形成微孔孔隙,但其孔结构不发达,且比表面积一般低于60 m2/g[19]。而污泥预处理或热解时添加活化试剂则能促进挥发性气体的释放,增加碳化产物的比表面积。王亚琛等[33]采用Fenton试剂活化污泥并联合高温热解,该过程强化了污泥在低温区的热解速率,从而提高了后期碳化产物的微孔体积。以化学试剂如ZnCl2、KOH或氧化性气体如CO2、水蒸气等与污泥混合制备高比表面积的碳化产物近年来在合成污泥基活性炭时被广泛使用。一方面,活化试剂可以加速污泥脱水,并在热解时形成含氢、氧等气体有利于碳化物成孔;另一方面,活化试剂能够抑制高温热解时产生焦油,防止堵塞孔径。表1所示为经不同活化试剂活化的污泥碳基催化剂样品,其比表面积均高于未活化时,且形成的样品孔径结构发达,并以介孔为主。
2.3 表面化学性质
表1 各种污泥碳基催化剂材料制备方法及其物理化学性质Table 1 A summary of the sewage sludge based carbon catalysts and their physiochemical properties |
| Catalyst | Preparation procedure | Method | BET surface area(m2/g) | Catalytic components | ref |
|---|---|---|---|---|---|
| FeSC | Dried sludge impregnated into 0.5 M FeSO4 solution and subsequently carbonized at 800 ℃ in the presence of N2 | Wetness impregnation | 14.3 | Magnetite, Quartz, Al2O3 | 19 |
| SC | Dried sludge carbonized at 800 ℃ in the presence of N2 | Direct pyrolysis | 57.6 | Carbon, ash | 14, 22 |
| SBC | Dried sludge impregnated into 3 M ZnCl2 solution and carbonized at 700 ℃ in the presence of N2 wash: 3 M HCl | Wetness chemical activation | 363 | Carbon | 23 |
| FMSAC | Carbonized sludge(by ZnCl2 pre-activation) was co-precipitated with Fe2+ and Fe3+ with NaOH addition | Chemical activation plus co-precipitation | 880~940 | Fe3O4, CaO, Quiz | 24 |
| R1 | Solid mixing of dried sludge with FeCl3(w/w=1∶1) and subsequently carbonized at 700 ℃ wash: 3 M HCl | Solid chemical activation | 517~836 | Fe species | 25 |
| szSAC | Dried sludge impregnated into the mixture of 3 M H2SO4 and ZnCl2 and then carbonized at 550 ℃ wash: 10% HCl | Wetness chemical activation | 179.9 | Surface-OH | 18 |
| szSAC/Mn | szSAC impregnated into KMnO4 solution and carbonized at 550 ℃ in the presence of N2 | Wetness impregnation | 3.7~11.7 | Mn(Ⅱ), Mn(Ⅲ), Mn(Ⅳ), surface-OH | 18 |
| DR-SA-A | Dried sludge was firstly carbonized with N2 and then activated with steam at 838 ℃, acid washed. | Steam activation | 497.4 | Dissolved organic matters and iron | 26 |
| nO x /SBAC FeO x /SBAC | Dried sludge was firstly activated with ZnCl2 and carbonized at 700 ℃ and had acid washed, then the carbonized products was impregnated into Mn/Fe solutions and re-carbonized at 600 ℃ | Chemical activation and wetness impregnation | 327~339 | Mn3O4, Fe3O4 | 15 |
| CFA/SC | Combined ZnCl2 activation and carbonization at 800 ℃ for the mixture of sewage sludge and fly ash(3 M HCl wash) | Chemical activation | 415 | Fe2O3, SiO2, Al2O3 | 27 |
| SS-Ti-700 | Combined hydrothermal reaction with TiOSO4 and carbonization at 700 ℃ | Hydrothermal reaction | 35.46 | TiO2, α-Fe2O3 | 28 |
| FAS-1-350 | Dried sludge impregnated into(NH4)2Fe(SO4)2 solutions, separated and calcined at 350 ℃ in the air | Wetness impregnation | 15.17 | α-Fe2O3, SiO2 crystallites | 29 |
| MC600 | Combined microwave digestion and KOH activation, then carbonized in the N2 | Chemical activation | 378 | O-containing groups, Fe3O4, α-Fe | 30 |
| SC-F-0.2 | Combined Fenton’s activation and carbonization at 600 ℃ | Radical activation | 46.3 | Fe3O4, α-Fe | 31 |
3 污泥碳基催化剂材料制备方法
污泥碳基催化剂的制备包括直接热解法、负载法、溶胶-凝胶法和水热合成法等。活化是热解制备多孔型碳基材料的重要手段,而通过负载、共混、表面改性等方法在碳基载体中引入不同的活性组分又可以实现碳基物料的催化功能[39]。这些活性组分可以是分散型过渡金属氧化物,如氧化铁、羟基氧化铁、氧化锰、二氧化钛等,也可以特指某些碳基表面官能团,如羧基、羟基、内酯基等。
3.1 直接热解法
早期的碳基催化剂制备是在不引入活性组分的前提下依靠污泥灰分热解形成的氧化物作为活性位点,并通过物理或化学活化增加碳基载体的比表面积来强化溶液相中活性物种和吸附质的迁移。决定催化剂活性的主要因素一般包括热解温度、热解气氛、活化剂种类和酸洗条件[32,39]等。其一般的制备流程包括:(1)剩余污泥在110 ℃左右脱水干燥;(2)机械粉碎并筛分;(3)活化剂活化处理并二次烘干形成污泥前躯体;(4)在惰性或活化气保护气氛中高温热解;(5)待冷却后的产物经洗涤后再次烘干、磨碎。常用的活化剂一般包括KOH、CaO、HNO3、ZnCl2或K2CO3,这类活化剂易引发污泥细胞脱水,强化热解形成挥发性气氛并促进表面成孔。
对采用直接热解制备的碳基催化剂而言,制备工艺对比表面积大小的影响直接决定了碳基物料对底物分子或氧化剂的吸附性能及其催化能力。Marques等[16]分别使用物理活化法(CO2、H2O)和化学活化法(K2CO3)制备污泥碳基催化剂,研究发现,不同活化方式制得的催化剂其催化活性与碳基载体的比表面积、灰分比例及表面pH均有较强的相关性。采用化学活化法制得的碳催化剂其S BET比表面积高达800 m2/g,苯酚和TOC的去除率可分别达到93.2%和82.1%,表明化学活化法对后期材料比表面积的增加和催化性能的提升有直接关系。
3.2 负载法
负载法是制备污泥基催化材料的主要方法,污泥中有机物在高温区形成碳基骨架,大量的无机灰分以多羟基氧化物和配合物存在并成为助催化剂,负载的活性金属则作为活性中心起直接催化作用。此外,许多无机变价金属在高温热解时催化污泥碳基表面形成含氧官能团,如—C 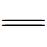 O、—COOH、—NH2等,这些官能团与变价金属结合形成更稳定的金属有机物骨架M—O—C,增强了催化剂稳定性,又可以通过电子传导增加金属表面的电子云密度,强化金属的催化活性[42]。根据被负载的对象不同,普通的负载法又可以分为碳化-负载-活化和负载-碳化的两段式和单段式工艺,常见的活性组分一般为FeCl3,Fe(NO)3,FeSO4,KMnO4和Mn(NO3)2等。
O、—COOH、—NH2等,这些官能团与变价金属结合形成更稳定的金属有机物骨架M—O—C,增强了催化剂稳定性,又可以通过电子传导增加金属表面的电子云密度,强化金属的催化活性[42]。根据被负载的对象不同,普通的负载法又可以分为碳化-负载-活化和负载-碳化的两段式和单段式工艺,常见的活性组分一般为FeCl3,Fe(NO)3,FeSO4,KMnO4和Mn(NO3)2等。
O、—COOH、—NH2等,这些官能团与变价金属结合形成更稳定的金属有机物骨架M—O—C,增强了催化剂稳定性,又可以通过电子传导增加金属表面的电子云密度,强化金属的催化活性[42]。根据被负载的对象不同,普通的负载法又可以分为碳化-负载-活化和负载-碳化的两段式和单段式工艺,常见的活性组分一般为FeCl3,Fe(NO)3,FeSO4,KMnO4和Mn(NO3)2等。3.2.1 两段式负载
两段式工艺是以强化污泥碳基载体在催化过程中的吸附作用为目的,利用前期活化剂对污泥的成孔作用制备出较高比表面积和发达孔径结构的碳基催化剂。热解温度和制备后样品的洗涤条件等将较大程度上决定催化剂的催化活性和金属稳定性。Zhuang等[15]用ZnCl2浸渍活化干污泥并热解制备碳基载体SBAC,再将载体浸渍在铁盐和锰盐配置的溶液中制成前躯体,并在600 ℃氮气保护条件下进行高温热解,待冷却后用盐酸进行反复洗涤。由于焙烧后载体中铁盐、锰盐等金属氧化物堵塞孔道,导致样品比表面积由398.6 m2/g分别下降至339.1和327.5 m2/g;但其负载金属氧化物与碳基物料在高温区相互作用并在表面形成羟基和酚羟基等特征官能团,可提高样品的化学等电点,有利于促进催化剂对水中阴离子污染物的吸附能力,强化降解物扩散到催化剂活性位点的速率。
3.2.2 单段式负载
近年来以过渡金属直接浸渍并活化干污泥或湿污泥的单段式负载工艺得到广泛应用。由于负载的活性组分本身会与污泥本体在高温热解时相互反应,影响碳基物料成孔、碳基表面含氧官能团形成以及活性组分特征氧化物晶体的生长[43],稳定性会得到提高,弥补了传统两段法制得的催化剂易失活、再生性能差的劣势。Sun等[44]用硝酸铁(磁源)及Mn、Cu、Co、Ni等不同金属盐溶液浸渍制备了磁性污泥碳,并用于催化臭氧体系降解布洛芬,结果显示,Mn掺杂的污泥碳催化活性最高,30 min内对布洛芬的降解率达86.2%。Mohedano等[45]将干污泥在FeCl3溶液中浸渍后再于管式炉中750 ℃热解得到污泥碳催化剂,由于制备过程使活性组分均匀分布,表现出较高的水相催化活性,对苯酚和乐果污染物达到65%的矿化度;铁浸出低,连续使用的稳定性好。
3.3 共混法
将干污泥与KOH、ZnCl2按一定比例进行固相混合是传统的制备高比表面积污泥活性炭的方法。Ros等[10]发现KOH混合干基污泥并热解后形成的碳其比表面积可达到1686 m2/g。近年来,为了开发出催化活性更高,活性组分分布更均匀以及催化剂稳定性更好的污泥基碳材料,以污泥与铁盐和含铁废弃物的直接固相混合活化技术得到了开发和应用。Bedia等[25]将好氧消化干污泥与氯化铁粉末按一定质量比固相混合后热解碳化,所得样品再经酸洗烘干后成型。他们发现当氯化铁与干污泥质量比达到3∶1时,活性氧化铁在载体上的分布更均匀,由于铁盐混合比例增加强化了其在高温区对污泥孔壁的腐蚀作用,导致冷却后碳基孔体积变大,平均孔径增加,后期酸洗更易脱除未附着在载体表面的铁氧化物,使催化剂的稳定性进一步增强。Zhuang等[27]以粉煤灰代替铁盐与污泥混合、活化并热解制备碳基多相Fenton催化剂。研究发现,混合粉煤灰后,碳基样品的比表面积可达415 m2/g,粉煤灰中大量的铁、硅、铝氧化物为固定污泥中重金属离子增加了吸附位点,使金属离子更容易在高温区掺杂进入氧化物晶格,从而大幅增加了催化剂的稳定性。
3.4 水热碳化法
水热法是指于压力容器内控制温度为150~350 ℃,使水成为电离常数较高的亚临界水并以此预活化污泥的一种热化学转化技术。水热碳化可以加速污泥中大分子有机物的水解和脱水,引发蛋白、木质素等解聚为低聚体或单体葡萄糖、果糖并逐渐转化为带有醛基的呋喃类衍生物,如羟甲基糠醛等[46]。这类化合物带有活泼的C 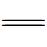 O键,极易与污泥中的活性金属如铁、钛、锌等结合,通过分子间缩聚形成新的带有催化活性中心的水热碳[47];同时,污泥中溶解的铁作为路易斯酸极易催化污泥水解并在水热碳基表面形成大量的含氧有机官能团,这些含氧官能团与金属活性物种具有极强的亲合力,能够抑制活性组分在载体表面发生团聚、降低活性组分的颗粒粒径、并强化活性组分在碳载体上的分布,阻止反应过程中活性物种在极端条件下向溶液相中的迁移[48],这种活性组分与碳基物料的化学吸附还可以促进物料在高温热解时的成孔作用,这将允许反应物在界面附近快速迁移至催化活性位点附近,从而提高催化剂的反应活性。
O键,极易与污泥中的活性金属如铁、钛、锌等结合,通过分子间缩聚形成新的带有催化活性中心的水热碳[47];同时,污泥中溶解的铁作为路易斯酸极易催化污泥水解并在水热碳基表面形成大量的含氧有机官能团,这些含氧官能团与金属活性物种具有极强的亲合力,能够抑制活性组分在载体表面发生团聚、降低活性组分的颗粒粒径、并强化活性组分在碳载体上的分布,阻止反应过程中活性物种在极端条件下向溶液相中的迁移[48],这种活性组分与碳基物料的化学吸附还可以促进物料在高温热解时的成孔作用,这将允许反应物在界面附近快速迁移至催化活性位点附近,从而提高催化剂的反应活性。
O键,极易与污泥中的活性金属如铁、钛、锌等结合,通过分子间缩聚形成新的带有催化活性中心的水热碳[47];同时,污泥中溶解的铁作为路易斯酸极易催化污泥水解并在水热碳基表面形成大量的含氧有机官能团,这些含氧官能团与金属活性物种具有极强的亲合力,能够抑制活性组分在载体表面发生团聚、降低活性组分的颗粒粒径、并强化活性组分在碳载体上的分布,阻止反应过程中活性物种在极端条件下向溶液相中的迁移[48],这种活性组分与碳基物料的化学吸附还可以促进物料在高温热解时的成孔作用,这将允许反应物在界面附近快速迁移至催化活性位点附近,从而提高催化剂的反应活性。 2Fe3+ + Fe2+ + 8OH- → Fe3O4 + 4H2O
值得注意的是,在水热活化阶段,污泥中大量的铁、铬、铝、铜、钙等金属离子从絮体解离并溶出,作为掺杂离子又进入二氧化钛、氧化铁等金属氧化物的晶格中,发生同晶取代。如铬离子和铁离子掺杂的锐钛矿型二氧化钛光催化剂具有更强的可见光吸收能力,而铁离子与硅氧化物形成的Si—O—Fe键则使催化剂具有更好的稳定性[46]。另外,污泥中的其他无机灰分和细胞残体则在后期碳化过程中转变为多孔模板,对提高催化剂对底物分子的吸附性能至关重要。Yuan等[51]以污泥为材料采用低温水解耦合高温热解的方法制备污泥SS-TiO2-700复合型光催化剂,研究发现球形TiO2粒状晶体团簇在以污泥为基质形成的模板上,污泥中与Ti原子尺寸相近的Cr和Fe原子作为施主和受主能级掺杂取代八面体中心的部分Ti原子,而Cu原子进入八面体晶格间隙中,使掺杂后的催化剂其禁带宽度由原TiO2的3.2 eV降低至2.8 eV;同时催化剂在N2吸附分压为P/P 0=0.6~1.0处陡峭上升说明催化剂中主要以污泥有机质燃烧、挥发和热解形成的大孔为主。
4 污泥碳基催化剂材料的表面改性
与活性炭类似,污泥碳基表面的化学性质对其催化能力、表面行为、亲疏水性和表面电势等均具有很大的影响,这些特性影响催化剂的吸附性和反应活性,决定催化剂在不同体系中的分散性与稳定性。通过对污泥碳晶进行酸处理、氧化处理调节污泥碳的表面化学性质、改变碳基官能团结构,尤其是含氧官能团如羧基、酸酐、内酯基等数量,从而达到强化催化剂表面缔合底物分子或氧化剂,加快反应速率的目的。
Yu等[52,53]比较了硫酸、盐酸和硝酸改性污泥碳基催化剂分别用于湿式催化氧化降解甲酚和异相Fenton反应去除氧氟沙星,表现出较高的降解效果,主要原因是酸性基团对污泥碳表面起到酸化作用,同时碳的含氧官能团被氧化为羧基和羰基,改性后的污泥碳的催化活性明显增强。虽然酸处理有利于洗脱灰分和增加催化剂孔体积和比表面积,然而接近沸点的洗脱温度易破坏催化剂碳晶的完整性,导致载体受腐蚀机械强度降低,影响后续的加工和使用。Wang等[54]改进了碳基的酸洗预处理方式,将挤出造粒后的碳化样品放置在0 ℃的低温硝酸溶液中进行浸渍,研究发现,低温酸处理可保持碳基材料颗粒的完整性,还会增加样品的比表面积;同时,低温改性保留了样品中的部分含氧碱性官能团如酚羟基、羰基和内酯基,这些供电子基团,可以加速Fe(Ⅲ)和Fe(Ⅱ)的电子转移,增加催化剂的催化活性。
5 污泥碳基催化剂在水环境催化领域的应用
5.1 催化H2O2的多相类Fenton反应
类Fenton法是在有催化剂存在的条件下利用过氧化氢产生的强氧化性自由基如·OH(E 0=+1.8~+2.8 eV vs NHE)氧化水中绝大多数有机物并使其矿化为二氧化碳和水,由于所用试剂无毒、绿色,处理效率快,不产生铁泥、操作pH温和等特点,成为难降解有机物在水中的主要处理技术之一[56]。为提高污泥碳基类Fenton反应氧化速率,克服活性组分易团聚、分散性差以及反应后难回收等问题,近年来对金属氧化物负载污泥基催化剂的研究不断深入。
5.2 基于过硫酸盐(PS)和过一硫酸盐(PMS)的催化降解
$≡M^{n+}+S_{2}O_{8}^{2-} → ≡ M^{(n+)+}+SO_{4}^{-}·+SO_{4}^{2-}$
Chen等[64]热解制备氧化铁负载厌氧消化污泥衍生生物碳。当热解温度从600 ℃升高到1000 ℃,Fe3O4逐渐转变为FeO,生物碳上负载的铁对二硫酸盐(PDS)活化和磺胺二甲嘧啶(SMT)降解的催化活性有较大影响。原位自由基清除和捕获测试表明,较高热解(1000 ℃)下催化体系中的主要活性氧(ROS)从·OH变化为$SO_{4}^{-}·$。此外,污泥碳催化材料可以通过协同促进反应物的吸附和界面处的C—O—Fe键的电荷转移来促进铁氧化物的催化活性。
Wang等[65]利用共沉淀法制备磁性铁负载污泥碳基催化剂催化过硫酸盐降解酸性橙7,研究表明污泥中活性组分Fe3O4和碳表面C  O官能团对PS的活化最为明显,利用甲醇和叔丁醇对自由基的淬灭试验证实,体系中同时存在·OH和S
O官能团对PS的活化最为明显,利用甲醇和叔丁醇对自由基的淬灭试验证实,体系中同时存在·OH和S  ·。进一步的研究发现,体系pH对基于吸附和两种自由基降解的反应历程的较大影响。这是因为,一方面酸性环境更有利于形成硫酸根自由基[66],而碱性环境易形成·OH 主导的催化体系(式(4));虽然·OH 比 S ·的氧化电位更高,但体系中同时生成的S 则存在与有机底物对自由基的竞争而降低自由基的氧化效率。另一方面,过渡金属负载的污泥碳基催化剂,其等电点一般介于中性偏碱,而绝大多数的阴离子染料易电离,因此弱酸性环境使得催化剂表面带正点而底物分子带负电,这种静电引力促进了底物分子在催化剂上的吸附,提高了底物分子向催化剂边缘的扩散速率。
·。进一步的研究发现,体系pH对基于吸附和两种自由基降解的反应历程的较大影响。这是因为,一方面酸性环境更有利于形成硫酸根自由基[66],而碱性环境易形成·OH 主导的催化体系(式(4));虽然·OH 比 S ·的氧化电位更高,但体系中同时生成的S 则存在与有机底物对自由基的竞争而降低自由基的氧化效率。另一方面,过渡金属负载的污泥碳基催化剂,其等电点一般介于中性偏碱,而绝大多数的阴离子染料易电离,因此弱酸性环境使得催化剂表面带正点而底物分子带负电,这种静电引力促进了底物分子在催化剂上的吸附,提高了底物分子向催化剂边缘的扩散速率。
O官能团对PS的活化最为明显,利用甲醇和叔丁醇对自由基的淬灭试验证实,体系中同时存在·OH和S $SO_{4}^{-}·+HO^{-}→ ·OH+SO_{4}^{2-}$
Li等[67]采用水热法合成MnFe2O4负载污泥活性炭催化过硫酸盐降解酸性橙,发现尖晶石结构变价金属Mn和Fe与S2 之间的电子转移加速了体系中S ·自由基的形成。苯酚的加入对过硫酸盐降解酸性橙起明显抑制作用,说明催化反应以表面催化为主,这与污泥碳晶边缘缺陷形成的羧基、羟基以及内酯基与过硫酸盐底物的电子转移有关:
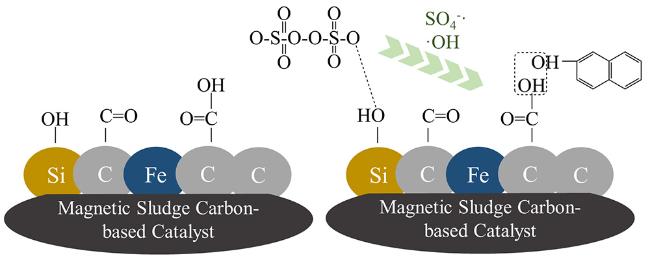
5.3 复合光催化反应
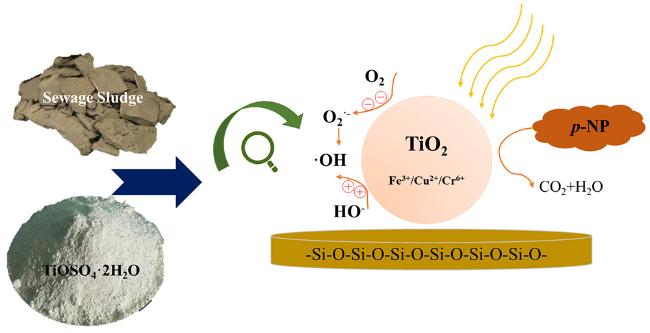
本课题组前期以污泥作载体,采用溶胶-凝胶法联用高温煅烧工艺将污泥与钛酸四异丙酯混合制备模板型TiO2可见光催化剂,污泥中挥发性有机质燃烧气化形成多孔模板,增加了复合材料的比表面积[77]。剩余无机物如钙长石、磷酸钙镁石复合TiO2形成如Ti—O—Si、Ti—C键增强了催化剂的稳定性。对样品受可见光照射后单电子氧空穴浓度进行ESR电子顺磁共振波谱分析发现,复合材料的空穴浓度远高于本征TiO2,形成的光生电子还原吸附位点附近的O2生成$O_{2}^{-}·$,进而生成·OH,通过自由基加成或电子转移引发—N  N—发色团断裂,并使染料褪色。
N—发色团断裂,并使染料褪色。
N—发色团断裂,并使染料褪色。 O2 + e- →$O_{2}^{-}·$
h + + OH- →·OH
h + + H2O →·OH + H+
UV/Fenton法是结合光催化和Fenton反应的一种新型高级氧化技术,具有很强的应用前景。反应体系在紫外光照射下直接催化过氧化氢产生羟基自由基并还原Fe3+,还原产物Fe2+与H2O2进一步反应生成·OH,从而加速体系的催化反应速率[78]。由于污泥中灰分在热解时能够形成多种类别的无机氧化物,某些无机过渡态金属氧化物能够吸收可见光产生瞬时电子并激发界面电子传递,诱导催化剂表面的还原反应。如Yuan等[28]使用富铁污泥碳化制备光催化剂,发现碳载体中的α-Fe2O3为光催化剂中的主要活性组分,这种催化剂在紫外或可见光激发下可以快速降解水中罗丹明B及对硝基苯酚,进一步的研究的表明,污泥中存在的大量无机氧化物MO被光激发后产生瞬时电子驱动界面电子转移还原氧化铁,诱发H2O2产生·OH和HO2·/ ·并降解有机物(图6),反应过程如方程式所示(其中M表示污泥碳基中的无机氧化物):
Fe(Ⅱ) +H2O2 → Fe(Ⅲ) + OH- + ·OH
$M+h\nu →M^{*}$
M* + Fe(Ⅲ) → Fe(Ⅱ) + M·+
Dye + hν → Dye*
某些染料受光照后产生敏化作用,形成激发态并诱导界面电子传递,如Tu等[79]使用铁负载的污泥碳基催化剂在H2O2和可见光存在下降解甲基橙时发现,染料受光照激发后可以快速传递电子还原  Fe(Ⅲ),加速
Fe(Ⅲ),加速  Fe(Ⅲ)催化H2O2产生羟基自由基。同时,Si-OH和Al-OH作为供电子基团,可以进一步被氧化断裂成羟基自由基。
Fe(Ⅲ)催化H2O2产生羟基自由基。同时,Si-OH和Al-OH作为供电子基团,可以进一步被氧化断裂成羟基自由基。
Fe(Ⅲ),加速 Fe(Ⅲ)催化H2O2产生羟基自由基。同时,Si-OH和Al-OH作为供电子基团,可以进一步被氧化断裂成羟基自由基。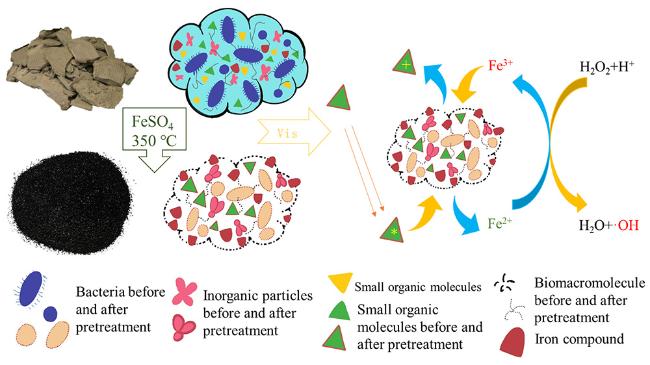
5.4 非均相催化臭氧氧化
催化臭氧氧化技术是在传统臭氧氧化的基础上发展起来的一种氧化效率高、反应速率快的污水深度处理技术。其中非均相臭氧氧化具有催化剂易回收、活性更高等特点。近年来,基于碳基的非均相臭氧氧化催化剂已经成为继金属氧化物、负载型催化剂和矿物型催化剂外的第四类主要催化剂[80]。由于活性炭易于吸附反应物与底物,因而大大加快了臭氧在催化剂表面的分解速率。有研究认为,吸附与臭氧氧化同时存在:首先,有机物被富集在活性炭表面,然后被同时吸附在碳基表面的臭氧催化分解,活性炭得到再生[81]。但也有观点认为,碳基表面缺陷形成的不饱和碳原子与表面吸附的水分子发生配位,使水电离形成羟基,与催化剂表面吸附的臭氧发生反应,生成羟基自由基等活性物种,氧化降解有机污染物[82]。这种表面羟基化作用也存在于某些过渡金属氧化物,如Al2O3、MnO x 、FeO x 等。污泥碳化产物由于具有可调控的比表面积能够加速有机物和臭氧的吸附,以及无机氧化物构成表面活性位点可以促进臭氧分解,近年来被当作非均相臭氧氧化催化剂被广泛研究。
Huang等[18]利用污泥制备MnO x 负载碳基催化剂并分析了其催化臭氧氧化草酸的性能,发现降解过程基于表面催化理论和溶液相自由基理论,即碳表面羟基诱导和锰氧化物还原O3的协同催化机理。首先,羟基与O3作用形成活性氧物种HO2·和 ·,这些含氧中间体进一步转移电子给O3并生成 ·。由于 ·不稳定,会迅速分解为O2和·OH并进入溶液相中攻击草酸分子;另外,变价金属Mn2+、Mn3+还原吸附在碳表面的O3,形成·OH,同时被吸附的草酸分子迅速还原氧化态的锰,其中间体进入液相被臭氧分子氧化。在反应过程中,溶液最佳pH值取决于催化剂的化学等电点pHpzc,中性表面羟基基团有利于自由基的产生,而过高或过低的溶液pH均会抑制表面羟基的臭氧催化作用[83]。
5.5 非均相催化湿式氧化
在湿式氧化体系(WAO)中加入适宜的污泥碳基催化剂,可使反应在更温和、更短的时间内完成,这种基于污泥为原料的非均相催化剂的开发受到了普遍关注,其催化过程影响因素、反应历程和催化剂的稳定性成为其中研究的焦点。Marques等[16,26]发现,碳基表面含氧官能团的类型和数量是引发体系自由基催化氧化的主要因素,以羰基为主的碱性含氧官能团通过界面电子传递与扩散至表面的氧气分子形成自由基。针对污泥碳基材料易失活等问题,Tu等[14]发现体系中形成的有机酸和盐酸等会诱导催化剂金属离子的溶出,尤其是铁、铜、钙等离子,虽然该过程促进了均相催化反应的进行,但大量金属离子的流出导致催化剂稳定性的下降,限制了污泥碳基催化剂的使用。
5.6 电化学催化氧化
污泥碳化后形成一类有机/无机复合产物,部分有机体或完全碳化形成碳基,受表面氧、氮原子对共轭电子的诱导产生极性;无机金属元素与灰分中SiO2形成稳定的Si—O—Fe、Si—O—C键,从而加了金属在碳晶载体上的稳定性和分散性,由于电化学反应本质上是一种电催化过程,电子通过外电路传递到粒子表面引发界面反应、电荷转移和氧化物种生成[88],因此,以污泥碳化形成的复合材料已经被证实在三维电化学系统中表现出较强的催化活性。
Hou等[89]探究了以市政污泥和铁泥混合制备铁负载碳基催化剂SAC-Fe,并考察其作为三维粒子填充电极降解洗煤废水的效率,发现该催化剂的活性和稳定性均优于颗粒活性炭和商业Fe3O4:灰分催化碳基形成的介孔提高了O2向碳基表面的扩散速率,加速了O2在粒子表面获得电子并被还原为H2O2的效率(式(14));污泥灰分中SiO2通过Si—OH解离在粒子周围形成微酸环境,诱导H2O2被负载的氧化铁分解为羟基自由基,将有机物分解和矿化。
O2 + 2e- + 2H+ → H2O2
Sun等[22]进一步验证了污泥碳基催化剂在三维电化学反应系统中降解酸性橙染料反应活性,研究发现过量粒子填充会破坏粒子间的绝缘性,使绝缘电阻下降、电流增加,造成短路;降解同时发生在液相和固相间,初期液相中底物的降解速率要明显高于固相反应并随反应时间增加而逐步降低。
6 污泥碳基催化剂的稳定性
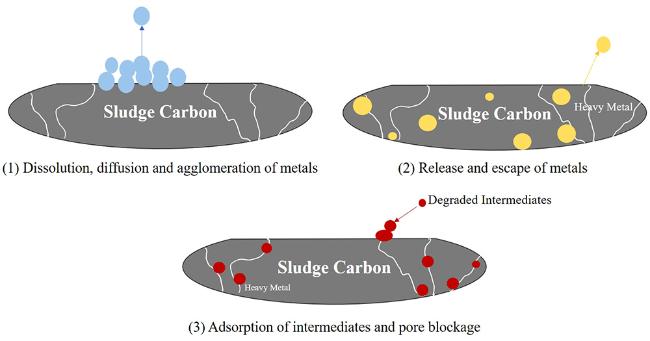
污泥碳基催化剂的稳定性和可重复利用性一直是污泥碳化材料大规模推广的关注点之一,因此寻求污泥中重金属等有害、有毒物质的沉积、钝化和固定,以及降低活性组分在降解过程中的溶解、逸出是目前该领域的研究热点。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
传统的金属负载型活性炭催化剂,其活性组分与污泥碳基间的结合力较差是影响催化剂稳定性的另一方面。催化时,降解过程产生的小分子有机酸,如草酸、乙酸等一方面降低体系pH,一方面与铁等金属络合,加速了活性组分的溶解。Zazo等[97]研究发现,采用该法制备的铁负载活性炭在多相Fenton中连续使用170 h后的铁逸出量可达到其初始负载量的50%。Rey等[98]证实Fe的逸出量与体系中产生的小分子酸浓度有关,对不同铁盐负载的活性炭在多相Fenton试验中的稳定性评价发现,铁的平均溶出量可达到45%。污泥形成的碳化产物无机灰分占比较高,这部分灰分在高温碳化时与活性组分,如铁等形成固溶体或产生特殊的化学键,如Si—O—Fe、Ti—O—Fe等,可以强化活性组分在污泥碳载体上的稳定性。
表2 几种污泥碳基材料在不同催化体系中的稳定性和可重复利用性比较Table 2 Comparison of stability and recyclability of several sludge carbonized materials in different catalytic systems |
| Leaching Species | Leaching Concentration | Reaction | Recyclability | ref |
|---|---|---|---|---|
| Fe | 0.6 g/L(2.5%of the total Fe load) | CWPO | 14.2% of the Fe load | 25 |
| Fe | not detectable | CWPO | 96% degradation efficiency obtained in third cycles of reaction | 92 |
| Fe | 0.037 mg/L(0.14% of total Fe loaded) Ca: 0.813 Cu: 0.029 Mg: 0.271 Zn: 0.027 | CWPO | 97% removal of AOII until at least 600 min | 19 |
| Fe | 1.9 mg/L for HNO3 treated SW 2.1 mg/L for H2SO4 treated SW 1.2mg/L for HCl treated SW 0.7 mg/L for SW | CWPO | 26.3% conversion of cresol for HCl-SW at 432 h 100% conversion of cresol for H2SO4-SW at 432 h 85.1% conversion of cresol for HNO3-SW at 432 h 40% to lower than 10% conversion rate for SW | 52 |
| Fe | 10.8 mg/L for HNO3 treated SW 11.7 mg/L for H2SO4 treated SW 0.8 mg/L for HCl treated SW 1.2 mg/L for SW | CWAO | Not mentioned | 7 |
| Fe | Fe: 18 mg/L Ni: 12 mg/L Zn: 4 mg/L Mn: 3 mg/L Cr: 3mg/L Mg: 2 mg/L | CWAO | For the fourth experiment, the differences after 4 h of reaction only amounted to 2.2% for phenol conversion and 9% for TOC conversion. | 26 |
| Fe | 27 mg/L(7% of the total Fe load) | CWAO | After 4 runs, the 2-CP conversion and the TOC removal were still very high. | 14 |
| Fe | 4.32 mg/L for SS-Fe-105 after 60 min 0.66 mg/L for SS-Fe-350 after 30min | Photo-Fenton | No obvious deactivation of the SS-Fe-350 catalyst in the six repetitive experiments was observed when compared with the first cycle. | 28 |
| Zn, Cu | 0.014 mg/L | PMS | The distributions of these heavy metals were unchanged though the MnO x /HCAS catalyst was reused up to 5 cycles. | 93 |
| Fe | pH 2.03:4.69 mg/L(2.14% of total iron) pH 3.01:3.06 mg/L | PS | three times for the oxidative degradation of AO7 | 65 |
| Fe | total Fe: 3.01 mg/L; Fe3+: 2.12mg/L | CWPO | It was observed that the mineralization rate decreased from 60.6 to 46.5% when the degradation rate of NOR decreased from 98.8 to 76.4%. | 94 |
为提高污泥碳基催化剂的稳定性,尤其是减少活性金属组分在溶液相中的逸出,对催化剂进行酸处理、在高温中热解以及通过预处理改变污泥前躯体与活性组分的作用是目前比较有效的控制手段。Marques等[26]比较发现,碳化后对催化剂酸洗并脱除灰分能有效降低反应过程金属的溶出率,同时提高焙烧温度,(>950 ℃)也有利于改善催化剂的稳定性。Yu等[7]进一步比较了盐酸、硝酸、硫酸不同酸后处理对催化剂稳定性的影响,发现盐酸更有利于增强催化剂的稳定性。Wen等[31]利用自由基预活化污泥,发现自由基攻击引起污泥菌胶团解离和细胞破裂,可大幅度降低污泥絮体颗粒粒径和表面Zeta电位,EPS的降解促使结合到污泥颗粒外表面的氧化官能团发生改变,充当了Fe阳离子的吸附位,促进铁离子在静电引力作用下被吸附到污泥表面,同时铁阳离子也起到凝结剂作用,将絮凝物聚集成大而致密的聚集体,并通过压缩双电层导致絮体脱稳、碰撞并聚合成包裹型的铁负载污泥前驱体,该方法可将铁在CWPO中的溶出率由原来的17.3%降低至2.77%。
7 结论及展望
市政污泥碳化合成功能型催化剂是实现高附加值污泥再生和资源化利用的重要途径。而污泥基碳质材料作为催化剂或载体在不同体系催化降解水中难降解有机物则具有以废治废,综合利用的特点。污泥碳化材料与普通活性炭相比具有更丰富的无机组成和表面官能团。基于不同工艺和制备方法所合成的催化剂具有较高的比表面积和电子迁移率,表面碳结构缺陷和其所含过渡金属氧化物能够激活多种氧化剂,如H2O2、PS、PMS、O3等并产生氧化性更强的氧活性物种;高温高压体系中溶出的无机离子诱导产生均相过氧化反应,加速金属与过氧化物之间的电子传递。污泥碳基中的多种无机灰分如SiO2、Al2O3、TiO2等具有助催化作用,可以形成Si—OH键形成催化剂表面微酸性环境或作为路易斯碱还原催化剂活性金属[81]。
污泥碳基材料凭借其自身的优异特性可以作为吸附材料、催化材料、半导体等多种功能性材料,其开发和应用具有典型的环境协调与兼容性特点,在溶液相催化降解体系中的应用具有巨大的潜力,这为污泥的处理和处置无疑提供了巨大的空间。尽管如此,该材料的应用尚处研究阶段,关于污泥来源对材料性能的影响、不同泥质对催化剂结构的调控机制、重金属参与的协同催化机制、碳基材料的安全性评价尚不完全明确,仍有许多关键问题亟待进一步探究。
(1)污泥的泥质受进水水质、水量、处理工艺不同而不完全相同,其有机质与泥沙含量等受季节影响波动较大[100],因此,对不同污泥来源,如市政、造纸、印染部门,或不同处理工艺,如厌氧消化、好氧消化、脱水污泥等对污泥碳化产物的组成元素、结构、催化性能等方面的影响有待更深入的研究。
(2)污泥碳基材料在催化体系中参与催化反应,受自由基或持久性过氧化物氧化而发生的表面结构改变对连续使用时催化性能的影响尚不明晰。已有报道显示催化过程中碳基表面受氧化而产生的表面极性基团对某些降解产物,如草酸等有特异性吸附[101],该过程对水质矿化程度的影响也有待进一步研究。
(3)制备过程中活化试剂种类与浓度、热解温度、升温速率、气氛等各种条件影响碳基催化材料的物理、化学性质,也影响其合成副产物如热解油、合成气的性质[32],如何控制反应条件优化各个产物的产出性质并实现全物料的综合回收也是污泥在碳化制备功能型催化材料方面尚待解决的问题。