




1 引言
2 生物矿化与仿生制备
2.1 生物矿化
2.2 生物矿物
2.3 仿生矿化

图1 (A)天然骨在纳米尺度上的结构水平; (B)将分子自组装、分子间交联和仿生矿化相结合的策略,制备类似于骨纳米结构的人工复合材料[43]Fig.1 Illustration of(A) the structural levels of natural bone at nanoscale and(B) the current strategy combining molecular self-assembly, intermolecular crosslinking, and biomimetic mineralization, to prepare artificial composite resembling bone nanostructure[43]. Copyright 2015, Wiley-VCH |
3 晶体成核
3.1 经典成核理论
3.2 非经典成核模型
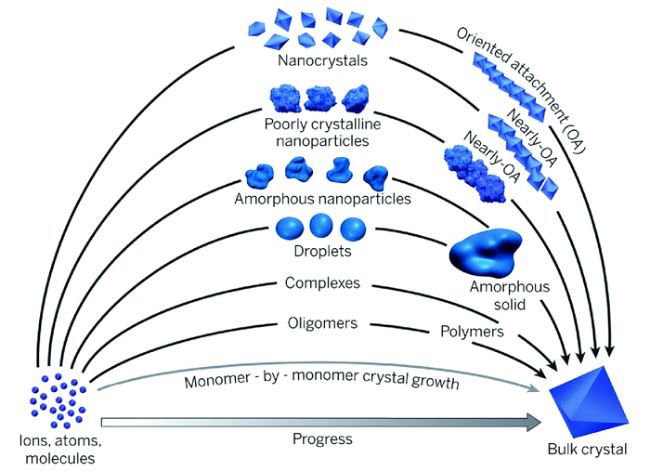
3.3 成核前驱体的认识
4 无机离子寡聚体与聚合
4.1 制备
4.2 结构
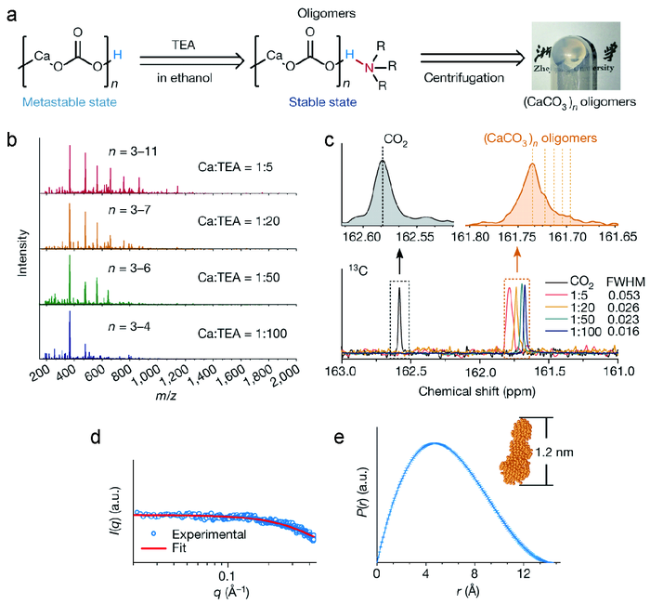
图3 (CaCO3)n寡聚体的制备和表征。(a)封端策略和制备凝胶状(CaCO3)n寡聚体的示意图;(b)不同Ca∶TEA摩尔比(CaCO3)n寡聚体的质谱图;(c)CO2或在乙醇中不同Ca∶TEA摩尔比的(CaCO3)n寡聚体的液体核磁共振碳谱图;(d)小角X射线散射测量的(CaCO3)n散射图;(e)(CaCO3)n寡聚体的对距离分布函数(P(r))[95] Fig.3 (a) scheme of the capping strategy and reaction conditions for producing gel-like(CaCO3)n oligomers;(b) Mass spectra of(CaCO3)n oligomers with different Ca∶TEA molar ratios;(c) Liquid-state 13C NMR spectra of CO2 or the carbonates of(CaCO3)n oligomers with different Ca∶TEA molar ratios in ethanol;(d) Scattering plots of(CaCO3)n measured by SAXS;(e) Pair-distance distribution function(P(r)) of the(CaCO3)n oligomers[95]. Copyright 2019, Springer Nature |
4.3 聚合交联
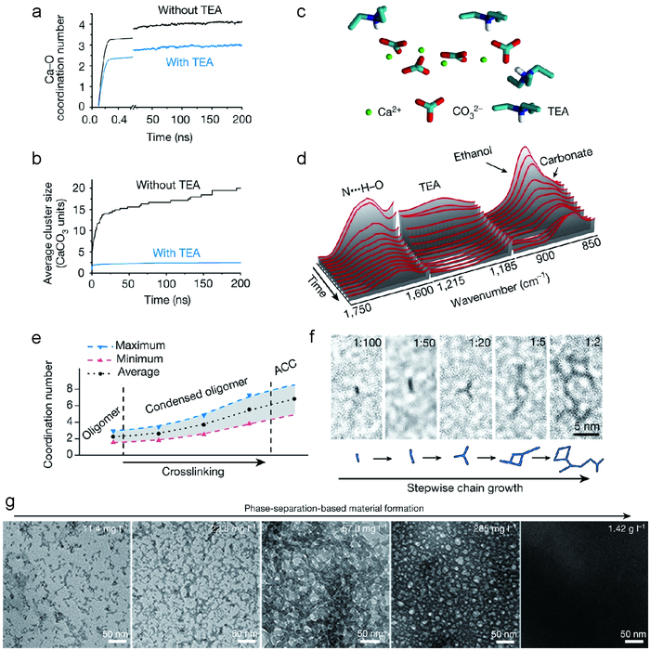
图4 (CaCO3)n寡聚体的可控交联。(a)Ca—O(来自碳酸根)配位数演变的分子动力学模拟;(b)平均团簇大小;(c)被TEA封端的典型的模拟CaCO3簇; (d)在(CaCO3)n寡聚体干燥过程中的原位傅里叶变换红外光谱;(e)交联过程中Ca—O配位数的变化;(f)不同Ca∶TEA比例(1∶100~1∶2)(CaCO3)n寡聚体生长的高分辨TEM图; (g)TEM图描述了(CaCO3)n寡聚体在聚合过程中向更大结构的转变[95] Fig.4 Controllable crosslinking of(CaCO3)n oligomers. (a) Molecular dynamics simulation of the evolution of the Ca—O(from carbonate) coordination number;(b) The average cluster size;(c) A typical simulated CaCO3 cluster capped with TEA;(d) In situ FT-IR spectra during the drying of(CaCO3)n oligomers;(e)The change in the coordination number of Ca—O during crosslinking;(f) High-resolution TEM images of(CaCO3)n oligomers grown at different Ca∶TEA ratios from 1∶100 to 1∶2;(g) TEM images depicting the transformation of(CaCO3)n oligomers to larger structures during condensation[95]. Copyright 2019, Springer Nature |
4.4 连续结构

图5 通过(CaCO3)n寡聚体交联制备无定形和类单晶CaCO3块体材料。(a)由(CaCO3)n寡聚体制备得来单块ACC的图;(b~e)扫描电镜(b,c)和透射电镜(d,e)图显示了制备的单块ACC的连续固相;(f)从单块ACC制备得到方解石的快照图;(g)制备单块方解石的偏振光光学显微镜图(POM);(h)结晶的单块CaCO3表面的SEM图;(i,j)结晶单块CaCO3的内部透射电镜图[95] Fig.5 Construction of amorphous and single-crystalline-like CaCO3 bulk materials by the crosslinking of(CaCO3)n oligomers. (a) Photograph of monolithic ACC prepared from(CaCO3)n oligomers;(b~e)SEM(b, c) and TEM(d, e) images indicating the continuous solid phase of the prepared monolithic ACC;(f) Snapshot of monolithic calcite prepared from monolithic ACC;(g) Polarized-light optical microscopy(POM) image of the prepared monolithic calcite;(h) SEM image of a surface on crystallized monolithic CaCO3;(i, j) TEM images of the inner bulk of crystallized monolithic CaCO3[95]. Copyright 2019, Springer Nature |
4.5 可塑制备
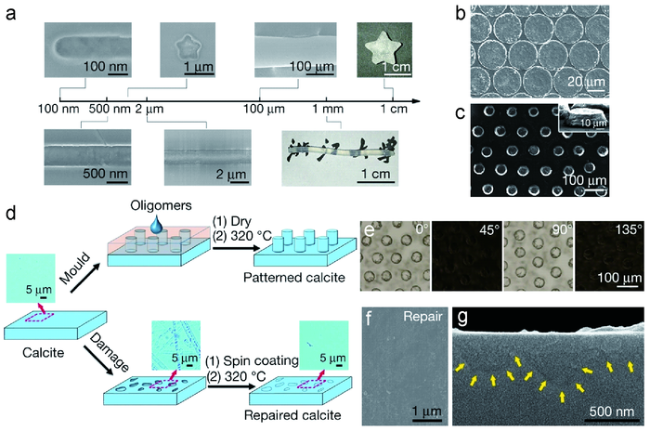
图6 通过使用(CaCO3)n寡聚体对CaCO3单晶材料进行可构建的工程化:(a)具有不同尺寸和形状的可塑CaCO3;(b,c)具有不同图案的可塑CaCO3;(d)在单晶方解石上图案构造的方案(顶部路径),以及将粗糙的单晶方解石修复成平滑的方解石的方案(底部路径);(e)以不同角度旋转的图案化方解石的POM图;(f,g)修复后的方解石的SEM图[95] Fig.6 Constructible engineering of CaCO3 single-crystalline materials by using(CaCO3)n oligomers. (a) Molded CaCO3 with different dimensions and morphologies;(b, c) Molded CaCO3 with different patterns;(d) Schemes for pattern construction on single-crystalline calcite(top path), and the repair of rough single-crystalline calcite to smooth calcite(bottom path);(e) POM images of the patterned calcite rotated at different angles;(f, g) SEM images of the repaired calcite[95]. Copyright 2019, Springer Nature |
5 基于无机聚合的仿生制备
5.1 组织修复

图7 牙釉质复杂结构的复制(A)酸蚀和修复后牙釉质的SEM图; (B)修复后牙釉质的三维AFM图; (C) (A)图中红色圆圈区域的高倍SEM图[103] Fig.7 Replication of the complicated structure of enamel. (A) SEM image showing both acid-etched enamel and repaired enamel;(B) A three-dimensional AFM image of repaired enamel;(C) High-magnification SEM image of the red circle in(A)[103]. Copyright 2019, AAAS |
5.2 有机无机共聚
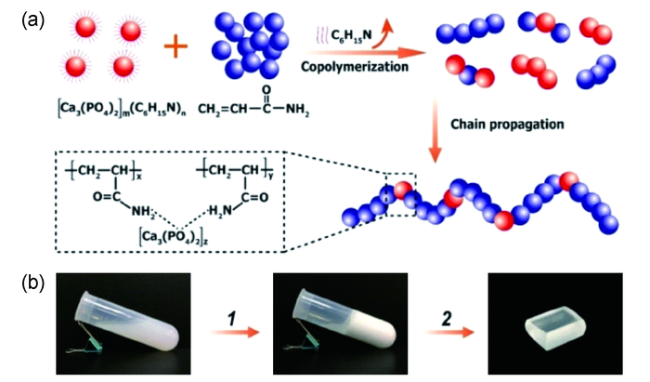
5.3 有机无机复合构建

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图9 (a)PVA/Alg/HAP混合微纤维的制备过程和网络微观结构的示意图;(b)PVA/Alg/CaP复合膜的光学照片;(c)PVA/Alg/CaP复合膜超薄部分的TEM图[112]Fig.9 (a) Schematic illustration of the preparation process and network microstructure of the PVA/Alg/HAP hybrid microfiber;(b) Optical photograph of the PVA/Alg/CaP hybrid film;(c) TEM image of the ultrathin section of the PVA/Alg/CaP hybrid film[112]. Copyright 2020, Wiley-VCH |