



Contents
1 引言
2 NRR催化过程
2.1 NRR热力学过程
2.2 NRR催化机理
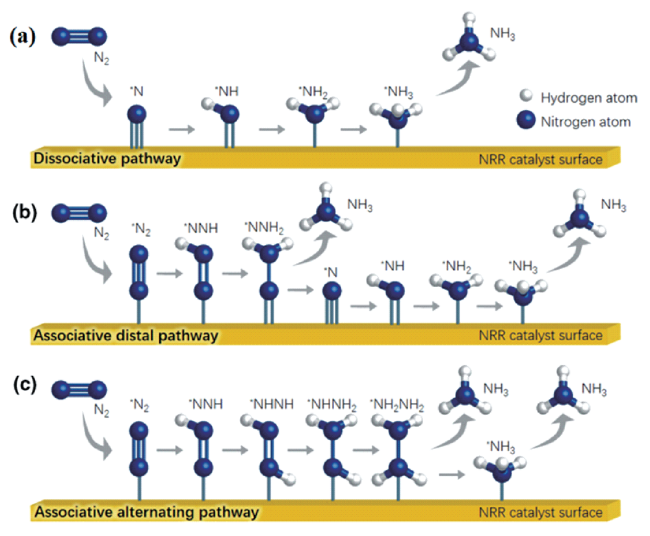
3 NRR非贵金属电催化剂
3.1 过渡金属氧化物
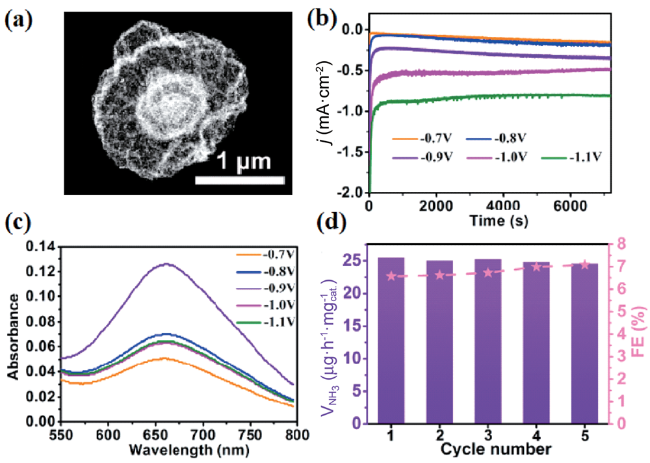
图2 (a) Cr2O3微米球高分辨透射电镜图;(b) 不同电压下Cr2O3微米球的i~t曲线; (c) 施加不同电压后电解液所对应的UV-Vis光谱图; (d) 稳定性循环实验[56]Fig. 2 (a) The HR-TEM image of Cr2O3 microspheres; (b) The i~t plots obtained from different applied potentials; (c) The corresponding UV-Vis spectra obtained after long-term tests; (d) The durability test for Cr2O3 microspheres[56] |
3.2 过渡金属氮化物

3.3 过渡金属硫化物
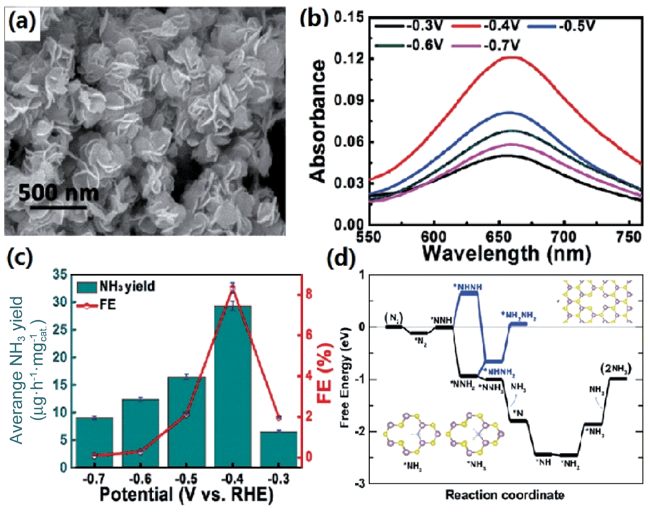
图4 (a) 富缺陷MoS2纳米花的扫描电镜图;(b) 施加不同电压后电解液所对应的UV-Vis光谱图;(c) 不同电压电解实验所对应的合成氨速率;(d) 基于富缺陷MoS2纳米花电催化NRR的自由能变化图[67]Fig. 4 (a) SEM image for defect-rich MoS2 nanoflower; (b) The corresponding UV-Vis spectra obtained after long-term tests; (c) The corresponding NH3 yields under different potentials; (d) Calculated free energy profile for NRR process on MoS2 basal plane[67] |
3.4 非金属催化剂
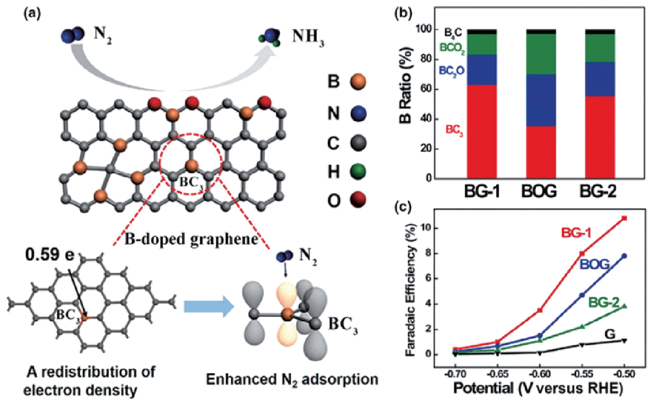
图5 (a) 基于B掺杂石墨烯电催化NRR结构示意图;(b) 不同B掺杂量的石墨烯;(c) B掺杂量对于石墨烯电催化法拉第效率的影响[76]Fig. 5 (a) Schematic illustration of NRR over B-doped graphene; (b) Percentages of different B doping configurations in three B-doped graphene samples; (c) The Faradaic efficiency values of BG-1, BOG, BG-2 and G at different applied potentials[76] |
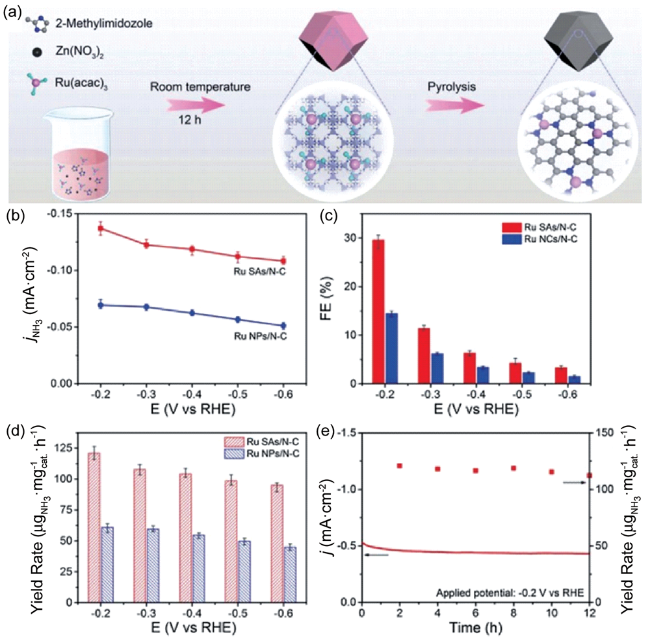
3.5 金属单原子催化剂
4 催化剂改性方法
4.1 催化剂晶面调控

图7 (a) 二十四面体Au纳米棒的原子层表面结构;(b) Au纳米棒几何模型及暴露的{730}面;(c) 电化学反应池构成;(d) Au纳米棒不同电压下的电催化NRR表现[91]Fig. 7 (a) Atomic level surface structures of Au THH NR; (b) Geometric models of an Au THH NR and exposed {730} facet; (c) Schematic for electrocatalytic NRR; (d) Yield rate of ammonia, hydrazine hydrate formation, and Faradic efficiency at each given potential[91] |
4.2 催化剂形貌与尺寸调控

4.3 缺陷控制

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图9 (a) 碳缺陷的碳纳米片(BCN)的扫描电镜图;(b) 基于理论模拟的BCN表面结构(灰(黑)、粉、红、黄和绿球分别代表C、B、O、N和H);(c) 不同催化剂对应的电催化NRR性能[97]Fig. 9 (a) The SEM image for BCN materials; (b) Schematic of the computational models. Gray (black), pink, red, blue, and green balls represent C, B, O, N, and H atoms, respectively; (c) The corresponding electrocatalytic NRR performance based on different catalysts[97] |
4.4 异原子掺杂
5 光催化NRR进展
6 NRR还原产物定量方法
表1 电催化剂性能对比Table 1 Summary of recently reported NRR electrocatalysts |
| Catalyst | Electrolyte | NH3 yield rate | Faraday efficiency(%) | Ref | |
|---|---|---|---|---|---|
| Noble metalcatalyst | Au nanocage | 0.5 M LiClO4 | 3.9 μg·h-1·cm-2 (-0.5 V vs. RHE) | 30.2 | 25 |
| Au nanorod | 0.1 M KOH | 1.648 μg·h-1·cm-2 (-0.2 V vs. RHE) | 4 | 91 | |
| Ru/C | 2 M KOH | 0.21 μg·h-1·cm-2 (-1.1 V vs. Ag/AgCl) | 0.28 | 27 | |
| RuPt/C | 18.36 μg·h-1·cm-2 (0.123 V vs. RHE) | 13.2 | 28 | ||
| Rh ultrathin nanosheet | 0.1 M KOH | 23.88 μg·h-1·m (-0.2 V vs. RHE) | 0.217 | 31 | |
| Pt/C | H+/Li+/N | 47.2 μg·h-1·cm-2 (1.2 V) | 0.83 | 32 | |
| Non-noble metal catalyst | Fe2O3/CNT | dilute KHCO3 solution | 0.22 μg·h-1·cm-2 (-0.2 V vs. Ag/AgCl) | 0.15 | 49 |
| Fe2O3-rGO | 0.5 M LiClO4 | 22.13 μg·h-1·m (-0.50 V vs. RHE) | 5.89 | 51 | |
| Fe2O3- x /CNT | 0.1 M KOH | 0.46 μg·h-1·cm-2 (-0.9 V vs. Ag/AgCl) | 6.0 | 52 | |
| Fe3O4/Ti | 0.1 M Na2SO4 | 5.6×10-11 mol·s-1·cm-2 (-0.4 V vs. RHE) | 2.6 | 53 | |
| Hollow Cr2O3 mircometer ball | 0.1 M Na2SO4 | 25.3 μg·h-1·m (-0.9 V vs. RHE) | 6.78 | 56 | |
| Mo2N | 0.1 M HCl | 78.4 μg·h-1·m (-0.3 V vs. RHE) | 4.5 | 62 | |
| VN | 0.05 M H2SO4 | 20.2 μg·h-1·cm-2 (-0.1 V vs. RHE) | 6 | 63 | |
| MoS2 | 0.1 M Na2SO4 | 8.08×10-11 mol·s-1·cm-2 (-0.5 V vs. RHE) | 1.17 | 66 | |
| Defect-rich MoS2 nanoflower | 0.1 M Na2SO4 | 29.28 μg·h-1·m (-0.4 V vs. RHE) | 8.34 | 67 | |
| MoS2/graphene | 0.1 M LiClO4 | 24.82 μg·h-1·m (-0.45 V vs. RHE) | 4.58 | 68 | |
| CoS x /NS-G | 0.05 M H2SO4 | 25.0 μg·h-1·m (-0.2 V vs. RHE) | 25.9 (-0.05 V vs.RHE) | 69 | |
| MoN nanosheet array | 0.1 M HCl | 3.01×10-10 mol·s-1·cm-2 (-0.3 V vs. RHE) | 1.15 | 92 | |
| Bi nanosheet array | 0.1 M HCl | 6.89 × 10-11 mol·s-1·cm-2 (-0.5 V vs. RHE) | 10.26 | 93 | |
| Bi4V2O11/CeO2 | 0.1 M HCl | 23.21 μg·h-1·mg-1 (-0.2 V vs. RHE) | 10.16 | 96 | |
| Metal-free catalyst | Oxidized carbonnanotube | 0.1 M LiClO4 | 32.33 μg·h-1·m (-0.4 V vs. RHE) | 12.50 | 73 |
| N-doped Carbon | 0.1 M KOH | 3.4×10-6 mol·h-1·cm-2(-0.4 V vs. RHE) | 10.2 | 75 | |
| B-doped graphene | 0.05 M H2SO4 | 9.8 μg·h-1·cm-2 (-0.5 V vs. RHE) | 10.8 | 76 | |
| N, P-codoped porous carbon | 0.1 M HCl | 0.97 μg·h-1·m (-0.2 V vs. RHE) | 4.2 | 77 | |
| BCN | 0.1 M HCl | 7.75 μg·h-1·mgcat.-1 (-0.2 V vs. RHE) | 13.79 | 97 | |
| nitrogen-decifient polymeric carbon nitride | 0.1 M HCl | 8.09 μg·h-1·m (-0.2 V vs. RHE) | 11.59 | 98 | |
| S-CNS | 0.1 M Na2SO4 | 19.07 μg·h-1·m (-0.7 V vs. RHE) | 7.47 | 99 | |
| single metal atom catalyst | Au/C3N4 | 0.05 M H2SO4 | 1.3 mg·h-1·m (-0.1 V vs. RHE) | 11.1 | 83 |
| Ru/N-C | 0.05 M H2SO4 | 120.9 μg·h-1·mg-1 (-0.2 V vs. RHE) | 29.6 | 84 | |
| Ru/N-C | 0.1 M HCl | 3.665 m ·h-1·m (-0.21 V) | 21 (-0.11 V) | 85 |