



Contents
1 Introduction
1.1 Porous noble metal nanostructures
1.2 Ordered porous noble metal nanostructures
1.3 Unordered porous noble metal nanostructures
2 Preparation of porous noble metal nanostructures
2.1 Hard-template method
2.2 Soft-template method
2.3 Bio-template method
2.4 Kirkendall effect
3 Application of porous noble metal nanostructures in biological detection
3.1 Localized surface plasmon resonance(LSPR)
3.2 Surface enhanced Raman spectroscopy(SERS)
4 Conclusion
1 引言
近年来,随着对贵金属纳米粒子的形貌、尺寸和晶面调控的深入研究,具有中空或多孔特征的一类贵金属纳米结构逐渐受到了人们的关注。相比于常规的实心纳米粒子,具有三维网状结构或孔状结构的纳米粒子最大的特点是具有低密度和高比表面积。而多孔的特性又赋予了它们与原子、离子和分子相互作用的能力,这种相互作用不仅能发生在多孔纳米粒子的表面,还能发生在纳米粒子的空隙之中[1]。此外,纳米颗粒之间孔结构的形成能够大大抑制颗粒的生长,减少了纳米粒子的聚集。因此,将贵金属纳米粒子的特殊性质与多孔结构相结合,能够得到具有更优越性能的新型材料。
1.1 贵金属多孔纳米结构
从纳米粒子的形貌有序度来分,可以将贵金属多孔纳米结构分为有序多孔纳米结构和无序多孔纳米结构。有序贵金属多孔纳米结构的特点是具有长程有序的周期性结构,如纳米孔阵列等[2],其在二维或三维尺度具有相同的结构单元,表现出很高的均匀度,性能重现性好。无序贵金属多孔纳米结构主要以单分散的纳米粒子为主,如多孔纳米花等[2],通过调控生长过程来改变其多孔特征,在形貌上具有很高的自由度,其结构具有多样性。有序和无序贵金属多孔纳米结构在实际应用的侧重点有所不同,有序结构多用于基底或固定的载体,无序结构多用于均相反应以及可移动的载体。因此根据不同的实际需求来选择设计多孔纳米结构,能够充分有效地发挥纳米粒子的潜在能力。
贵金属多孔纳米结构的设计和合成为新型结构在应用领域的发展拓宽了道路。在催化领域,多孔贵金属纳米材料可以作为催化剂或催化剂的载体。例如利用多孔Au、Ag、Pt结构制备的催化剂,能够广泛应用在CO氧化、开环反应、交叉偶联反应[4,5,6]等方面。在这些催化反应中,多孔的贵金属纳米粒子表现出比常规纳米粒子更高的催化活性、效率和稳定性。此外,通过优化多孔粒子的尺寸,贵金属多孔纳米结构在能源和生物医学领域也表现出很大的应用潜力。例如通过设计微尺度或纳米尺度的贵金属多孔单元,发挥其高吸附率、高孔隙率、高导电性和小尺寸效应,在燃料电池[7]、药物递送[8]、细胞成像[9]、表面增强拉曼光谱(SERS)[10]和病毒消除[11]等领域具有广泛的应用。多孔结构可以大大扩展活性位点,增强燃料分子的质量跃迁,从而提高其在燃料电池应用中的活性和耐久性。同样,多孔或空心结构可以提供更大的载药能力和多通道给药。此外,多孔结构可以产生粗糙的表面,从而产生高密度的“热点”,提高SERS性能。
1.2 有序贵金属多孔纳米结构

本课题组在有序贵金属多孔纳米结构的设计合成方面也取得了一些进展,如Rao等[16]利用SiO2胶体晶作为模板,利用吸附在硅球表面的Au颗粒作为晶种,H2O2作为还原剂,将外加的氯金酸还原形成SiO2/Au的核壳结构,最后用HF酸去除模板,制备了有序多孔金纳米碗阵列。
1.3 无序贵金属多孔纳米结构
与有序贵金属多孔纳米结构相比,无序贵金属多孔纳米结构的形式更加多样化,应用范围也更加广泛。从这个层面来说,设计和组装新型无序贵金属多孔纳米结构并提高金属的催化和光谱性能尤为重要。无序贵金属多孔纳米结构从形貌和尺度上主要可分为两类:一类是与有序多孔纳米材料类似,具有很高的尺寸和结构单元,但是孔洞没有周期性结构,表现为混乱孔分布;另一类是指单分散的贵金属纳米粒子,每个纳米粒子表现出空心多孔的特性。

2 贵金属多孔纳米结构的制备
目前,模板法是制备有序贵金属多孔纳米结构最有效的方法之一,根据使用模板的不同可分为硬模板法、软模板法和生物模板法,而所使用模板的结构和性质也会影响所得有序多孔纳米结构的特性。常用的硬模板主要包括自组装的胶体晶、介孔硅球[24]、沸石分子筛[25]和氧化铝膜[26]等。这些模板多包含三维有序结构或互相连通的介孔,可以被广泛地用来合成各种贵金属及贵金属合金的有序多孔结构,并能有效地调控其结构和形貌。而通过使用液体晶或各种有机聚合物等作为软模板,亦可以简便、温和地制备出各种有序多孔贵金属结构[27,28,29]。此外,利用自然界存在的各种具有多级结构的生物材料作为模板,也可以制备出许多不同结构的有序多孔纳米材料。与硬模板和软模板相比,生物模板成本更低、无毒、环境友好并大量存在于自然界中[30]。最重要的是,生物模板本身就具有非常独特的多级结构和形貌,为制备多级孔复合材料提供了条件。
2.1 硬模板法
硬模板法主要是以有序排列的无机物胶体晶为模板,在其表面沉积相应贵金属前驱体溶液,再经过高温退火、萃取或强酸腐蚀等方法除去模板,最后得到具有多孔结构的贵金属纳米材料。硬模板法因模板的稳定性高且其光学性质的高度可调成为目前使用最多的制备有序贵金属多孔纳米结构的方法。其合成路线主要包括三个步骤:首先制备并组装出具有特定形貌的有序/无序模板;然后将通过原位定向沉积、共沉积、核-壳结构的组装等过程将贵金属纳米粒子沉积在模板表面或间隙之中;最后移除最初使用的模板,得到所需的多孔贵金属产物[31]。通过组装法得到的长程有序的SiO2或PS微球阵列是最为常用的硬模板。利用Stöber法可以方便地制备出尺寸从10 nm到几个微米级的SiO2微球,使用这些尺寸不同的模板能够制备出各种不同孔径的多孔贵金属纳米材料。此外,硅球表面众多的Si-OH基团可以与许多不同的有机基团如—NH2,—SH和—COOH进行键合,从而根据不同的需要或制备方法得到不同的功能化多孔纳米材料。Colvin等[32]利用SiO2纳米晶作为模板,较早发展了一种通用的制备贵金属有序多孔结构的方法。首先通过垂直沉积法将巯基改性的胶体硅组装在玻璃片表面,然后胶体Au通过吸附作用和渗透作用非电镀地沉积在胶体硅的缝隙中,在热处理去除硅球模板后,即得到有序多孔的Au纳米阵列。
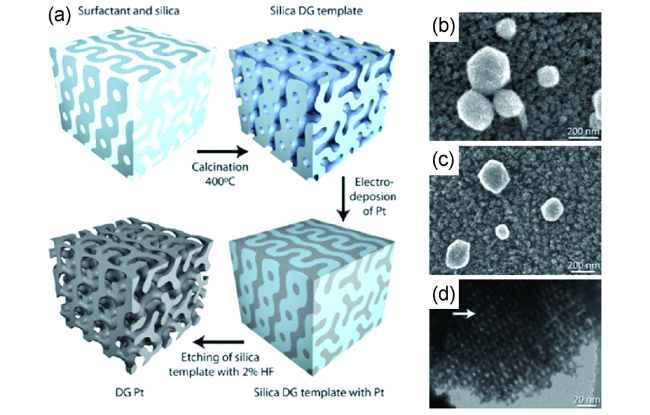
Kibsgaard等[33]以在玻璃碳盘上生长的介孔二氧化硅膜作为模板,利用电沉积的方法制备了一种双陀螺形的Pt纳米结构,见图3。首先在HCl和乙醇的条件下,将正硅酸四乙酯预水解,然后将含有EO19-PO43-EO19的乙醇溶液加入体系中形成涂液,EO19-PO43-EO19在体系中起到表面活性剂的作用。然后将抛光的玻璃碳盘浸入至涂液中,在50%的湿度下通过浸涂(dip-coating)的方法将涂液均匀地沉在玻璃碳盘表面。此时在表面活性剂的诱导下二氧化硅膜会自组装形成双陀螺形结构,待溶剂挥发后,在400 ℃下煅烧4 h,即得到介孔的二氧化硅模板。最后通过电沉积法将Pt沉积到模板上,再用2 wt%的HF刻蚀30 min去除二氧化硅模板,最终制备得到了一种双陀螺形的多孔Pt纳米结构。

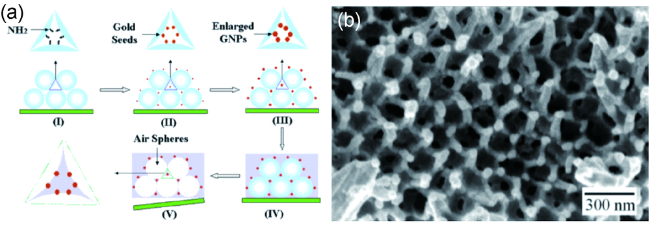
然后在聚苯乙烯球表面通过沉积的方法覆盖一层Au膜形成PS/Au结构,将另一种尺寸较小的聚苯乙烯球仍按照上述方法单层有序地铺在PS/Au结构上方。利用沉积的Au作为电极的一极,通过电化学方法将Ag纳米粒子沉积在胶体晶表面,最后移除聚苯乙烯球,即得到了这种有序多孔的Ag超结构。这种制备方法新颖高效,产物的形貌均匀,具有很高的普适性,不仅可以用来制备相似结构的Au、Ag、Ni等单金属,还可以用来合成金属氧化物如MnO2或有机聚合物如聚吡咯的有序多孔超分子结构。
本课题组的Ding等[35]提出过一种制备具有强局域表面等离子体共振(LSPR)效应的有序多孔聚苯乙烯/金纳米结构的方法。如图5,首先通过垂直蒸发法将有序SiO2胶体晶组装在玻片上,然后将金纳米粒子通过静电作用吸附在硅球表面,得到有序的SiO2/Au阵列。接着将聚苯乙烯的甲苯溶液灌注在胶体晶内,待甲苯挥发完全后,聚苯乙烯会将SiO2/Au固定住,且包覆在SiO2/Au阵列表面。将聚苯乙烯膜轻轻揭下,利用HF将SiO2腐蚀掉,即可得到有序多孔的PS/Au结构。通过控制硅球的大小和厚度可以调控产物孔径的大小和厚度,且实验参数的不同会对金纳米粒子的LSPR峰位置产生影响,可以通过这种结构实现目标分析物的非标记检测。
硬模板法是一种最为常见的模板制备贵金属多孔结构的方法,模板的选取较为多样化,贵金属的尺寸可通过所选取模板的大小来进行有效地调节。但也存在着一些缺点:(1)不同模板表面的电荷、基团、亲疏水性等各不相同,因此根据模板的不同需要设计不同的制备路线,制备方法较为繁琐;(2)模板最终需要通过高温、酸腐蚀等方法进行移除,在这个过程中,如何维持贵金属多孔纳米结构的形貌,是硬模板法需要解决的一个重要问题。
2.2 软模板法
软模板法是以具有软结构的有机分子或有机分子聚合物为模板,通过自组装和相分离过程产生多级前驱体结构,最后除去模板即可得到具有多级孔结构的纳米材料。与二氧化硅、沸石等硬模板相比,有机分子聚合物的成分、尺寸、形貌、渗透性等特性能够很方便地进行调控,表现出丰富多彩的结构多样性。
两亲性分子,例如表面活性剂或两亲性嵌段共聚物,均包含亲水部分和疏水部分,其通过共价作用连接在一起。当混合液的浓度超过临界胶束浓度时(critical micelle concentration,CMC),两亲分子会自组装形成球形结构。这种表面活性剂浓度的增加会促使溶致型液晶相(lyotropic liquid crystals, LLCs)的形成。LLCs具有较长的空间周期性结构和结构多样性,晶格参数在2~15 nm[36],可以作为一种理想的软模板来合成贵金属多孔纳米结构。Attard等[37,38]首次提出并发展了通过LLCs来制备多孔Pt纳米结构的方法,随后一系列特异形貌的贵金属多孔纳米结构利用这种方法合成出来,目前已成为制备贵金属多孔纳米结构的常用方法。
Yuan等[39]利用聚合物P(DEAEMA-co-O-B-EG)作为模板,制备出了一种三维带状的多孔金纳米结构,见图6。首先利用十八烷、聚乙二醇等有机分子的聚合反应制备出聚合物P(DEAEMA-co-O-B-EG)。然后直接将其与氯金酸混合,利用抗坏血酸作为还原剂,控制反应时间,直接还原制备了这种新颖的多孔金纳米结构。这种金纳米材料是由许多放射状的片状结构组装形成的,每个纳米片的厚度约为2 nm。实验结果表明产物的形貌与聚合物的分子量有关,通过改变PEG的分子量(400~600),还能使产物整体架构从圆柱形转变为梭形。Yamauchi等[40]利用溶致型液晶相(LLCs)的二嵌段共聚物作为模板,通过电沉积的方法制备出了一种新型的三维介孔Pt纳米结构,孔径大小约为15 nm。Lee等[41]利用双连续表面活性乳胶通过还原银离子法得到了多级孔结构的块状银。

软模板法因其模板的特殊性,通常用来制备无序贵金属多孔纳米结构。与硬模板法相比成本较低,操作简单且环境污染较小,但是具有合成过程较难控制、孔径大小不能精准调节等方面的缺陷。
2.3 生物模板法
生物模板法主要是以自然界存在的多孔生物材质作为模板,通过将贵金属纳米结构沉积在生物材料上来制备一类多孔的贵金属功能材料。常用的生物材质包括动物硬组织如贝壳和植物纤维等。
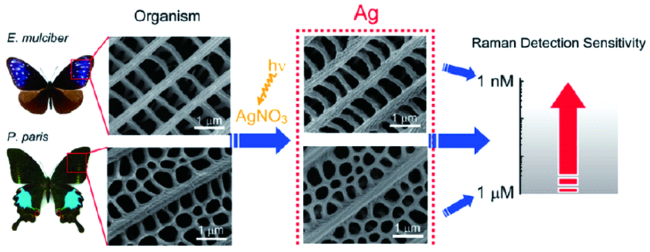
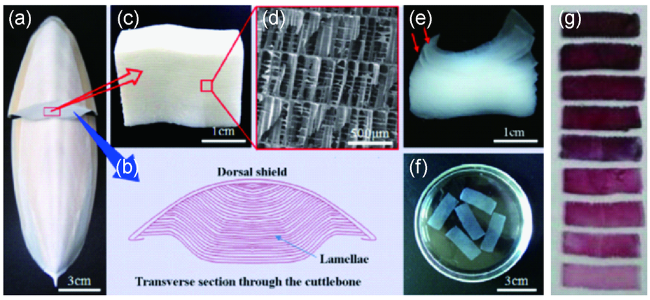
生物模板法成本较低且环境友好,天然的生物有机质能够提供有效的多孔基底,通过直接沉积贵金属可以简单快速地制备多孔贵金属纳米材料。但这种方法具有一定的缺点:(1)生物有机质模板的结构是自然形成的,对多孔的一些特性如孔隙率、孔径等无法进行调控;(2)生物材料的个体差异性使得不同批次产物的贵金属沉积量难以保持一致,如何保持实际应用中样品的性能再现性仍是亟需解决的问题。
2.4 柯肯达尔效应
利用伽伐尼(Galvanic)置换反应与柯肯达尔(Kirkendall)效应来制备单分散的贵金属多孔纳米结构是目前多孔材料领域的一个重要的方法。在这个过程中仍需要使用模板,但与硬模板法稍有区别的是,使用的模板会作为反应物在多孔贵金属结构形成的过程中逐渐被刻蚀掉。常用的模板为具有还原性的金属或金属氧化物如Ag、Cu、Cu2O等。此外,模板本身即为形貌、尺寸、组成可控的纳米结构,因此通过这种方法能够制备出结构更加多样化、种类更加繁多的多孔贵金属纳米材料。
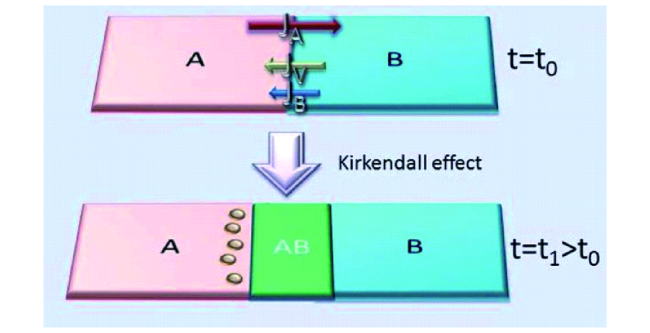
在反应过程中,通常以一种金属或金属氧化物作为模板,较高氧化还原电位的金属盐作为前驱体。在Galvanic置换反应和Kirkendall效应的作用下,模板会将贵金属前驱盐还原,并在模板表面沉积。而模板的表面会被逐渐刻蚀并形成空洞,最终得到贵金属(或合金)中空多孔材料。Ag相对于Au、Pt、Pd来说具有更低的氧化还原电位,通常被作为一种理想模板来合成各种形貌的中空贵金属多孔纳米结构。Puntes等[46]通过控制Galvanic反应和Kirkendall效应同时发生,提出了一种不同形貌贵金属及其合金多孔纳米结构的制备方法。通过严格控制反应和扩散过程,合成了圆柱形、球形、立方Au,Au-Ag,Au-Ag-Pd等金属中空纳米结构。反应过程分为两个阶段,即首先在Ag表面形成Galvanic孔洞,Au原子由于电位差在其表面沉积;然后表面的Au原子与内部的Ag原子之间通过扩散耦合效应形成Kirkendall中空结构,见图10。Xia等[47]利用Ag纳米立方作为模板,通过各种不同的方法制备出了多孔Au纳米笼,并系统地研究了其LSPR效应、药物负载与释放、光热治疗等性能。
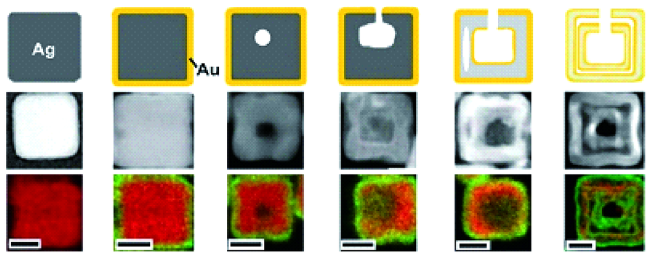

利用柯肯达尔效应和伽伐尼置换反应能够对产物的形貌、组分、生长晶面等进行精确地调控,多用于制备单分散的多孔贵金属纳米粒子。通过不同氧化还原电位的贵金属之间的相互反应,可以制备出一系列单金属、多金属的多孔纳米材料。但模板的使用成本较高,反应过程需要精确控制,对实验操作的要求较高。

3 贵金属多孔纳米结构在生物检测中的应用
多孔贵金属纳米材料特别是Au、Ag纳米结构具有独特的LSPR或SERS等光学性质,目前在生物传感检测上具有非常广阔的应用前景。此外,多孔结构还能够负载更多的生物分子,在分子检测、药物输送和释放等方面也具有潜在的应用价值。
3.1 局域表面等离子共振(LSPR)
贵金属纳米粒子表面的自由电子与入射光之间的相互作用会产生共振现象,即局域表面等离子共振(LSPR)。对于金、银、铜等纳米粒子,其谐振峰主要出现在可见光区;而Ru、Rh、Pd、Pt等贵金属粒子的谐振峰主要出现在紫外光区。然而,这些贵金属纳米粒子的LSPR峰位置能够随着粒子的尺寸、组成、形貌和晶体电子密度的不同而发生变化,甚至偏移至近红外区或红外区。Marks等[52]指出,贵金属纳米晶的粒径在50~350 nm之间为LSPR峰的振动距离,即“等离激元长度”(Plasmon length)。此外,由于贵金属纳米晶的形貌不同,其LSPR峰的数目可以出现多个。如金纳米棒可以在纵向产生一个较长波长的共振,在横向可以产生一个较短波长的共振。这两种模式可以通过控制纳米棒的纵横比和长度来进行调控[53]。多孔贵金属纳米粒子在形成过程中由于其介电常数在三维空间内呈多样性变化,因而可以产生非常有趣的LSPR特性。如在相同的尺寸下,中空的Au纳米框具有比实心Au纳米粒子更为灵敏的LSPR峰变化,实验和理论也证明这种Au纳米棒的光学灵敏度与其壁长和壁厚的比例有关[54]。通过与待检测分子的相互作用导致的LSPR峰的变化,能够实现生物分子的高灵敏检测。
Zhang等[55]利用立方体金纳米笼形成过程中LSPR峰的变化实现了H2O2的高灵敏检测。利用H2O2对AuAg合金纳米粒子进行刻蚀,随着Ag被逐渐刻蚀,最终会形成Au纳米笼,而在这个过程中会伴随着产物LSPR峰的偏移。通过加入H2O2量与LSPR峰偏移量和强度之间的关系,可以实现H2O2的检测。实验结果表明这种方法对H2O2的检测线性范围为5×10-2~5×10-7 M。Hong等[56]设计了一种基于SPR原理的、以有序多孔Ag为基底的生物分子检测方法。利用这种有序多孔Ag的反射峰在添加生物素标记的牛血清蛋白后发生红移,对抗生物素蛋白的检测可达到100 pM。Liu等[57]总结了具有优异LSPR特性的一系列Au纳米粒子的制备方法,并对在保证Au纳米粒子光学特性的同时,如何经过合理的表面修饰实现Au纳米粒子在生物传感、生物成像等方面的实际应用进行了深入的探讨。
3.2 表面增强拉曼光谱(SERS)
表面粗糙的Ag和Au具有很强的拉曼增强性能。这些贵金属多孔纳米结构表面粗糙,其多孔特征使其具有许多缝隙或凸起从而形成热点,因此可以作为很好的SERS基底材料。有序多孔金、银阵列由于其长程效应,可作为一种均匀的SERS基底,且在此基础上制备出的具有有序多孔结构的金、银复合纳米材料能具有更大的比表面积及“热点”效应,其SERS增强效果可进一步提高。而无序多孔Au、Ag纳米结构其多元化的形貌和高比表面积,可以将SERS技术与其他应用互相结合,扩展SERS技术的应用领域。
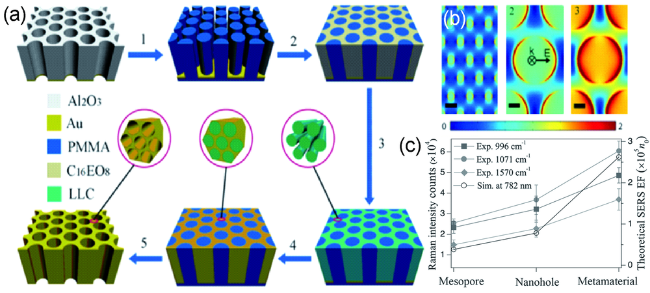
图13 (a)三维有序多孔Au纳米结构的制备过程,(b)产物表面的归一化电场振幅分布,(c)不同峰位置处的拉曼强度和电磁增强因子模拟[58]Fig. 13 (a) Synthesis procedure of 3D ordered porous Au nanostructures,(b) Normalized electric field(E-field) amplitude distribution at the surface,(c) Raman intensities at different peaks and the simulated EM enhancement factors[58] |
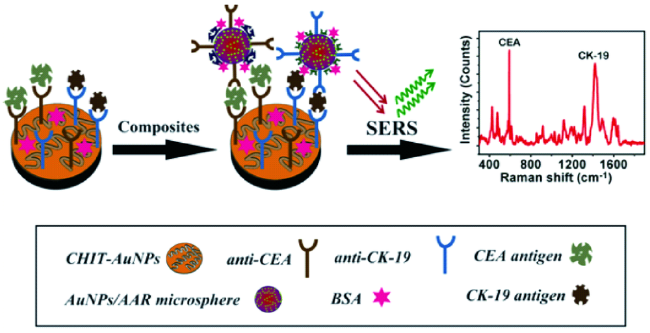
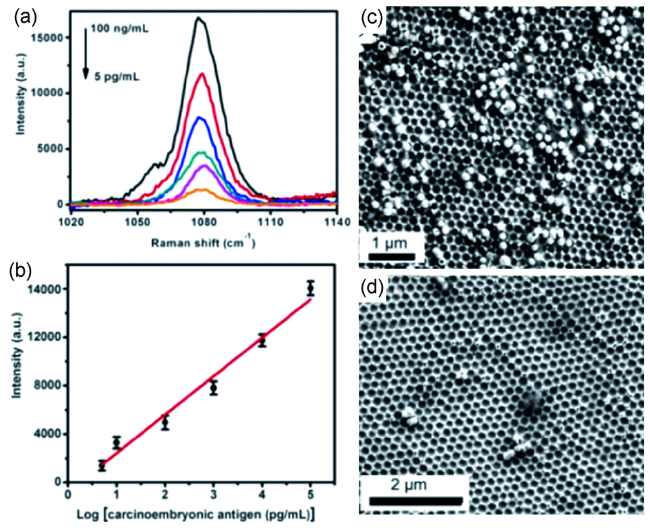
本课题组一直致力于SERS在生物传感领域的研究。Lu等[60]制备了一种具有多孔微通道的水杨酸树脂微球,然后以此为基底,在壳聚糖保护下,通过静电吸附与配位作用结合金纳米粒子,得到了一种微孔水杨酸树脂微球/Au纳米结构。然后用这种Au结构固定抗体和标记分子,利用anti-CEA(Coating)和anti-CK-19作为捕获探针,构建了一种检测肺癌标志物CEA和CK-19的生物免疫传感器,实现了SERS与电化学的双重检测,如图14。结果表明这种生物传感器在SERS模式下CEA和CK-19的检测限分别为0.62和1.01 ng/mL。这种超灵敏可靠的检测肺癌标志物的方法为肺癌的早期诊断提供了新的思路。Li等[61]首次将有序金纳米碗阵列应用于癌症标志物的检测研究领域,通过在三维有序金纳米碗上修饰anti-CEA作为捕获基底,在金纳米壳上吸附4-MBA作为拉曼信号分子,通过捕获抗体、标记抗体和待检测癌坯抗原CEA形成“三明治”结构,实现了对肺癌标志物CEA的SERS灵敏检测,如图15。在最优化的条件下CEA的检测范围5~100 pg/mL,最低检测限是1.73 pg/mL,且在这个浓度范围内SERS强度与CEA浓度之间有良好的线性关系,实现了癌症标志物的灵敏定量检测。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论
贵金属多孔纳米材料因其特定的形貌、多元化的组分、高的比表面积、优异的光学特性等在先进功能材料的不断发展中占有着重要的地位。目前利用各种模板法能够精确有效地调控贵金属多孔纳米材料的尺寸、孔径、形貌等,但制备过程仍较为复杂、应用成本也较高且难以大批量地合成。此外,在生物检测中,如何对纳米材料进行合理的表面修饰,提高光学性质的稳定性和生物相容性,是贵金属多孔纳米材料在实际应用中所面临的挑战。但随着对纳米结构越来越精细的调控,更多具有最优化孔径、比表面积、空间结构的新型多孔贵金属功能纳米材料正被逐渐制备出来。相应地,具有更高选择性、高稳定性和更高检测灵敏度的生物传感检测方法也有望被开发。贵金属多孔纳米结构在催化、生物传感、癌症诊断等方面会越来越具有实际应用的潜力和价值。