




Contents
1 引言
2 水相加氢反应
2.1 水相加氢反应的类型
2.2 溶剂水的作用
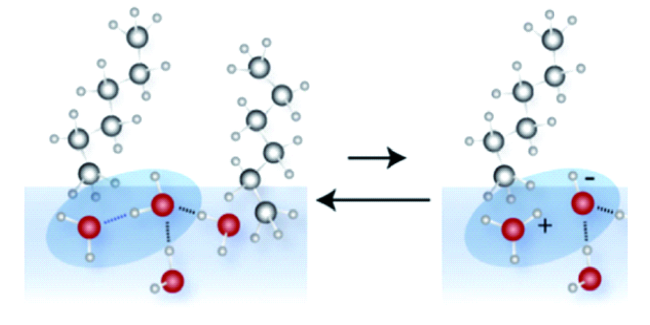
图1 水/烃(油)界面处模拟水分子的示意图。深灰色球:C原子,浅灰色:H原子,红色:O原子,虚线:氢键。透明矩形描绘了水和碳氢化合物之间的界面[14]Fig. 1 Schematic illustration of the simulated water structure at a water/hydrocarbon(oil) interface. Color key: dark gray spheres=C, light gray=H, red=O. Dotted lines are hydrogen bonds. The transparent rectangle depicts the interface between water and the hydrocarbon[14] |

图2 部分离解水在Ru(0001)上的扫描隧道显微镜(STM)图像:(a)145 K沉积后完好的水条带,(b)145 K退火30 min后部分游离的H2O-OH条带。DFT优化结构中,OH基团中的O原子用橙色标记[16]Fig. 2 Partial dissociation of H2O on Ru(0001): STM images of(a) intact water stripes after deposition at 145 K and its transformation to(b) partially dissociated H2O-OH stripes after 30 min annealing at 145 K. In the DFT optimized structures shown, the OH group is highlighted by an orange O atom[16] |
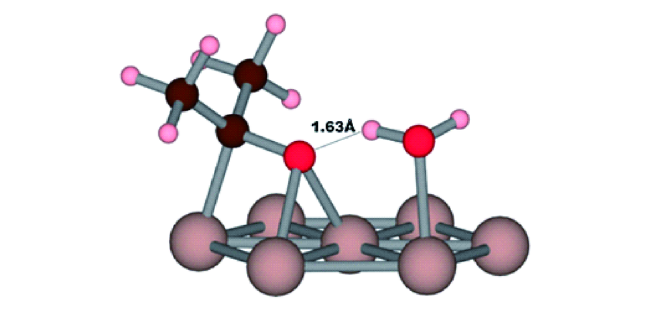
图3 丙酮和水共吸附在Ru(0001)上。丙酮与水形成氢键的吸附构型(0.18 eV), 深棕色球=C,粉红色=H,红色=O,浅棕色=Ru[22]Fig. 3 Acetone and water co-adsorbed to Ru(0001). This adsorption configuration of acetone is stabilized by 0.18 eV through formation of a hydrogen bond(depicted with annotated line). Dark brown spheres=C, pink=H, red=O, light brown=Ru[22] |
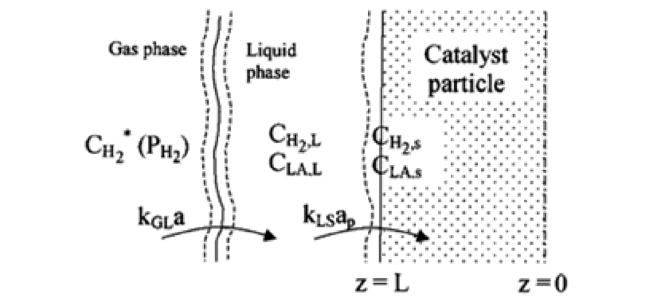
2.3 水相加氢反应的传质
3 水相加氢催化剂
3.1 加氢反应催化剂的类型
表1 水相加氢反应中各种催化剂的活性总结Table 1 Summary of catalytic performance of various catalysts in aqueous phase hydrogenation |
| Type of catalyst | Catalyst | Substrate | Reaction condition | Conv./% | ref |
|---|---|---|---|---|---|
| noble metals | Ru-MC-g | benzoic acid | H2(4 MPa), 120 ℃, 2 h | 94 | 35 |
| Rh/H-Beta | diphenyl ether | H2(4 MPa), 120 ℃, 3 h | 80 | 37 | |
| Pt/H-Beta | diphenyl ether | H2(4 MPa), 120 ℃, 3 h | 64 | 37 | |
| Ru/H-Beta | diphenyl ether | H2(4 MPa), 120 ℃, 3 h | 70 | 37 | |
| Ru/Al2O3 | levulinic acid | H2(2 MPa), 50 ℃, 1 h | 22 | 49 | |
| Ir/CNT | levulinic acid | H2(2 MPa), 50 ℃, 1 h | 96 | 49 | |
| Ru/CNT | levulinic acid | H2(2 MPa), 50 ℃, 1 h | 65 | 47 | |
| Ru/C | guaiacol | H2(4 MPa), 250 ℃, 2 h | 56 | 50 | |
| Rh/C | guaiacol | H2(4 MPa), 250 ℃, 2 h | 15 | 50 | |
| Pt/C | guaiacol | H2(4 MPa), 250 ℃, 2 h | 2 | 50 | |
| Pd/C | guaiacol | H2(4 MPa), 250 ℃, 2 h | 0 | 50 | |
| Pd/C | phenol | H2(4 MPa), 250 ℃, 2 h | 82 | 50 | |
| Ru/CNT | cellobiose | H2(5 MPa), 185 ℃, 3 h | 88 | 51 | |
| metal oxides and metal composites | 4%Rh-MoOx/SiO2(Mo/Rh=0.13) | levulinic acid | H2(6 MPa), 80 ℃, 6 h | 100 | 38 |
| 4%Ir-MoOx/SiO2(Mo/Ir=0.13) | levulinic acid | H2(6 MPa), 80 ℃, 6 h | 100 | 38 | |
| 4%Ru-MoOx/SiO2(Mo/Ru=0.13) | levulinic acid | H2(6 MPa), 80 ℃, 6 h | 100 | 38 | |
| 4%Rh-MoOx/SiO2(Mo/Rh=0.13) | lactic acid | H2(6 MPa), 80 ℃, 6 h | 78 | 38 | |
| Pt-ReOx/C | sorbitol | H2(6.21 MPa), 245 ℃, WHSV(2.92 h-1) | 99 | 41 | |
| Pt-ReOx/Zr-P | sorbitol | H2(6.21 MPa), 160 ℃, WHSV(0.16 h-1) | 92 | 42 | |
| Pd1Fe3/Zr-P | sorbitol | H2(6.21 MPa), 245 ℃, WHSV(2.92 h-1) | 16 | 44 | |
| Pd/WOx/-Al2O3 | guaiacol | H2(7 MPa), 300 ℃, 150 min | 100 | 52 | |
| non-noble metals | Raney Ni | levulinate esters | H-donor(2-PrOH), room temperature, Ar, 2 h | 87 | 40 |
| 20%Cu/ZrO2-OG(oxalate-gel) | levulinic acid/ formic acid | formic acid, N2(1 MPa), 180 ℃, 5 h | 60 | 41 | |
| 5 wt%Ni-HAP | levulinic acid | H2(0.5 MPa), 70 ℃, 4 h | 18 | 53 | |
| 10%Ni/Al2O3 | levulinic acid | H2(3 MPa), 200 ℃, 3 h | 29 | 54 | |
| 7.9 mol%Co/AC | vanillin/formic acid | formic acid, N2(0.5 MPa), 180 ℃, 4 h | 6 | 45 | |
| Co@NC-700 (7.9 mol%Co) | vanillin/ formic acid | formic acid, N2(0.5 MPa), 180 ℃, 4 h | 96 | 48 | |
| Fe@NC-700 (7.9 mol% Fe) | vanillin/ formic acid | formic acid, N2(0.5 MPa), 180 ℃, 4 h | 10 | 48 | |
| Ni@NC-700 (7.9 mol% Ni) | vanillin/ formic acid | formic acid, N2(0.5 MPa), 180 ℃, 4 h | 37 | 48 | |
| Cu@NC-700 (7.9 mol% Cu) | vanillin/ formic acid | formic acid, N2(0.5 MPa), 180 ℃, 4 h | 4 | 48 | |
| 4Co/Al2O3(nCo/nAl=4) | levulinic acid | H2(5 MPa), 180 ℃, 3 h | 6 | 55 |

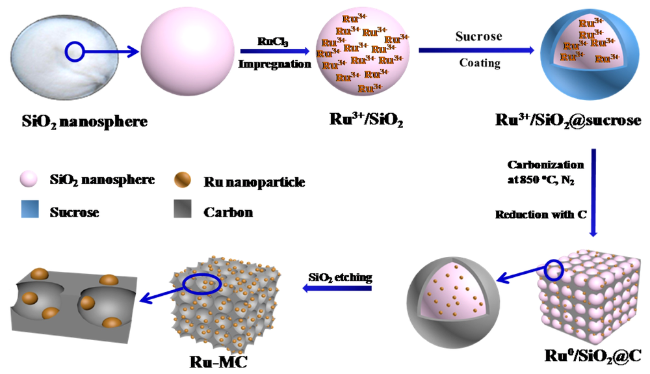
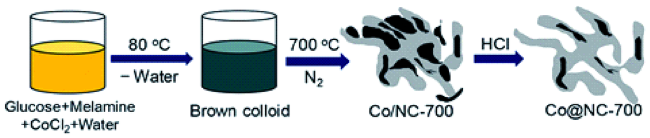
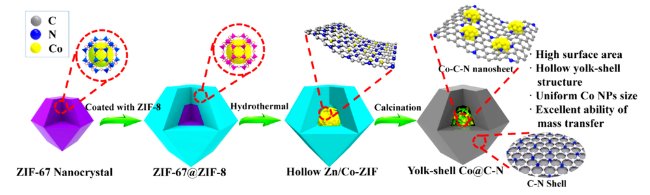
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}