




Contents
1 引言
2 胺基配体单茂金属催化剂
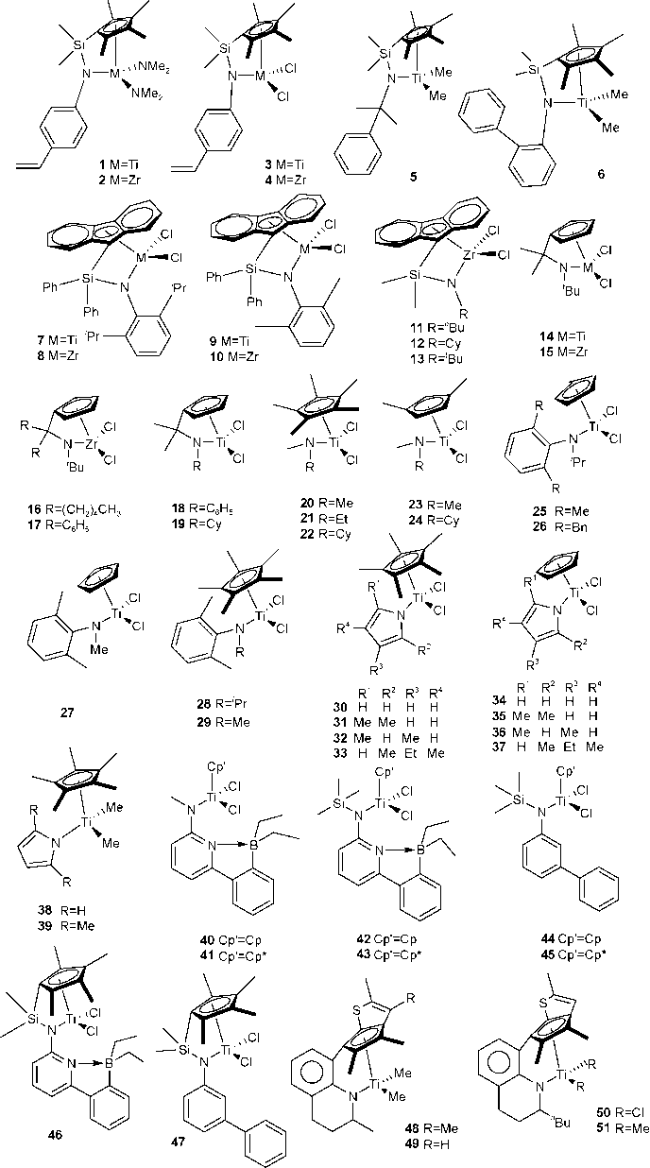
表1 催化剂25~29的乙烯聚合[24][a]Table 1 Ethylene polymerization by 25~29[24][a] |
| Cat. | T (℃) | Al/Ti mol. ratio | Activity[b] | 10-4 | (℃) |
|---|---|---|---|---|---|
| 25[e] | 20 | 200 | 316 | 371.5 | 137.9 |
| 25[e] | 35 | 200 | 330 | 105.5 | 136.5 |
| 26[e] | 20 | 200 | 340 | 383.5 | 137.7 |
| 26[e] | 35 | 200 | 354 | 138.8 | 136.9 |
| 27[e] | 20 | 200 | 220 | 325.4 | 136.8 |
| 27[e] | 35 | 200 | 231 | 103.3 | 136.4 |
| 28 | 20 | 200 | 1852 | 94.1 | 138.9 |
| 28 | 35 | 200 | 1974 | 70.6 | 138.1 |
| 28 | 50 | 200 | 1270 | 55.9 | 139.0 |
| 28 | 35 | 300 | 1692 | 83.5 | 139.5 |
| 28 | 35 | 400 | 1594 | 68.3 | 140.1 |
| 29 | 20 | 200 | 1806 | 96.8 | 139.3 |
| 29 | 35 | 200 | 1924 | 82.1 | 139.0 |
| 29 | 35 | 300 | 1856 | 58.4 | 137.9 |
[a] Polymerization conditions: 70 mL toluene, 2×10-6 mol catalyst, 1.5 B/Ti molar ratio, 15 min, 5 bar ethylene pressure.[b] Activity in kg-PE/mol-Ti-h.[c] Measured in decahydronaphthalene at 135 ℃.[d] Determined by DSC at a heating rate of 10 ℃min-1. The data from the second scan have been used.[e] 4×10 -6 mol. |
表2 催化剂25~29的乙烯-己烯-1共聚合[24][a]Table 2 Ethylene-hexene-1 copolymerization by 25~29[24][a] |
| Cat. | 1-Hex (mol·L-1) | Activity[b] | Incorpor.[c] (mol %) | ×10-4 | pdi[d] | rErH[e] |
|---|---|---|---|---|---|---|
| 25[f] | 1.5 | 142 | 9.5 | 15.72 | 3.41 | - |
| 26[f] | 1.5 | 152 | 10.0 | 10.42 | 3.62 | 0.547 |
| 27[f] | 1.5 | 72 | 8.7 | 10.04 | 3.53 | - |
| 28 | 0.5 | 3364 | 18.4 | 26.33 | 2.42 | 0.485 |
| 28 | 1.5 | 3608 | 31.8 | 20.14 | 2.55 | 0.393 |
| 29 | 1.5 | 1610 | 35.9 | 17.45 | 3.31 | 0.437 |
[a] Polymerization conditions: total 70 mL of toluene+1-hexene, 2×10-6 mol catalyst, 200 Al/Ti molar ratio, 1.5 B/Ti molar ratio, 15 min, 35 ℃, 5 bar ethylene pressure.[b]Activity in kg-Poly/mol-Ti-h.[c] Calculated based on13C NMR spectra.[d]Measured by GPC analysis.[e] rErH =4[EE][HH]/[EH+HE]2.[f] 4×10-6 mol. |
| Cat. | Activity (kg-PE/mol-Ti-h) | Mn×10-3 | pdi |
|---|---|---|---|
| 30[a] | 5550 | 22.7 | 2.4 |
| 31[a] | 1500 | 20.7 | 2.4 |
| 32[a] | 1740 | 22.8 | 2.3 |
| 33[a] | 1950 | 21.8 | 2.3 |
| 34[a] | 330 | 2.18 | 2.2 |
| 35[a] | 750 | 2.61 | 2.1 |
| 36[a] | 930 | 2.91 | 3.6 |
| 37[a] | 1410 | 3.17 | 1.7 |
| 38[b] | 10 020 | 112 | 2.5 |
| 39[b] | 8100 | 160 | 2.8 |
Reaction conditions: toluene total 30 mL, ethylene 6 atm, 10 min, 25 ℃, 100 mL scale autoclave,[a] d-MAO(prepared by removing AlMe3 and toluene from MAO), Al/Ti=15 000.[b] [iBuAl]/[Ti]=250, [[Ph3C][B(C6F5)4]]/[Ti]=3.0. |
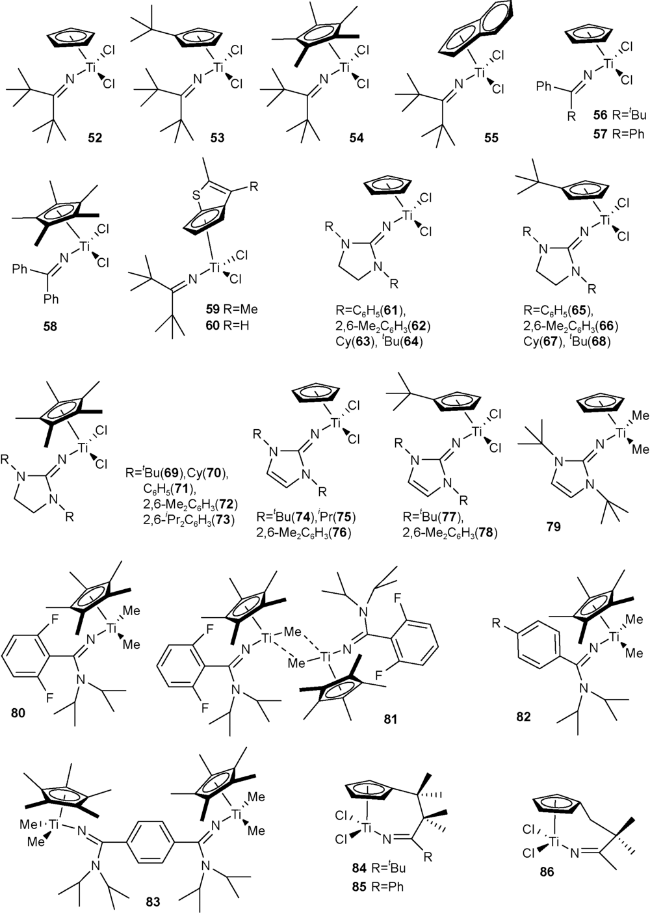
3 亚胺基配体单茂金属催化剂
4 膦亚胺基配体单茂金属催化剂
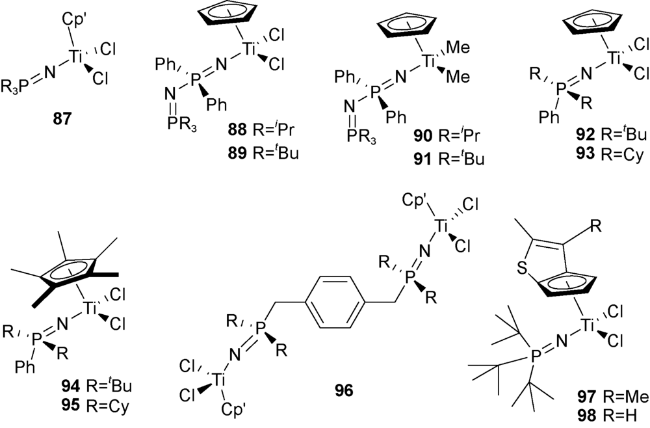
表4 催化剂97、98的乙烯-辛烯-1共聚合及其与Cp*Ti[NP(tBu)3]Cl2的比较[38][a]Table 4 Comparison for ethylene-octene-1 copolymerization by 97, 98 and Cp*Ti[NP(tBu)3]C12[38][a] |
| Cat. | Activity[b] | Incorpor. (mol %)[c] | Mw ×10-3 | pdi |
|---|---|---|---|---|
| 97 | 9.0 | 8 | 543 | 2.44 |
| 98 | 8.0 | 8 | 752 | 2.17 |
| —[d] | 23 | 5 | 204 | 3.32 |
[a] Polymerization conditions: 30 mL toluene solution of 1-octene(0.30 M, 1.0 g), 1.0 mmol Ti, 4.0 mmol [Ph3C][B(C6F5)4], 0.20 mmol AliBu3, 60 psig ethylene, 3 min, 80 ℃ initial temperature.[b] Activity in unit of 106 g/mol-Ti h.[c] 1-Octene content in the copolymer determined by the1H NMR.[d] Cp*Ti[NP(tBu)3]Cl2 was used as catalyst. |
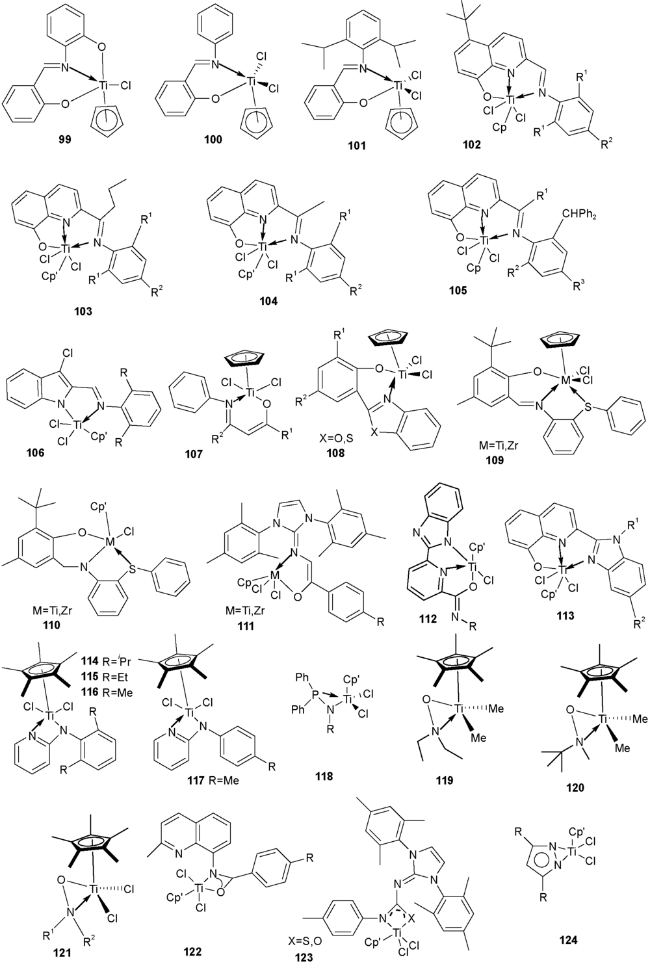
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}