




Contents
1 Introduction
2 Syntheses of pillararene-based mechanically interlocked molecules
2.1 Syntheses of pillararene-based rotaxanes
2.2 Syntheses of pillararene-based catenanes
2.3 Syntheses of pillararene-based mechanically selflocked molecules
3 Molecular mobility of pillararene-based mechanically interlocked molecules
3.1 Shuttling mobility of pillar[5]arene-based [2]rotaxanes
3.2 Shuttling mobility of pillar[6]arene-based [2]rotaxane
3.3 Stretching/contraction mobility of pillar[5]arene-based [c2]daisy chain
4 Functionality of pillararene-based mechanically interlocked molecules
4.1 Chirality inversion
4.2 Förster resonance energy transfer
4.3 Stimuli-responsive gel
4.4 Langmuir film
4.5 Rotaxane-branched dendrimers
5 Conclusion and outlook
1 引言
大环主体的设计合成与功能化推动着超分子化学的发展,冠醚[1]、环糊精[2]、葫芦脲[3]、杯芳烃[4, 5]等大环分子的发展依托于相对应的主客体化学及其应用。柱芳烃[6,7,8]是一类新型大环主体分子,包含柱[5]芳烃至柱[15]芳烃等成员,其中以柱[5]芳烃最易于合成和衍生化。由于柱芳烃具有柱状富电子空腔,其衍生物可与多种客体分子如烷烃、氰基衍生物、胺基衍生物、咪唑、吡啶盐、铵盐、芳香化合物等发生识别作用形成主客体络合物[9,10,11,12,13,14,15],进而构筑如轮烷、索烃等机械互锁结构[16],并在质子通道[17]、药物传输与释放[18,19,20]、光学探针[21]、气体吸附[22]、生物成像[23]、异构体分离[24]等领域具有广泛应用。
机械互锁结构(mechanically interlocked molecules)[25]是一类分子间基于非共价键作用而相互缠绕锁定的超分子体系,由于其拓扑美学结构与分子内相对运动的特点而备受化学研究者青睐。机械互锁结构包括轮烷[26]、索烃[27]、分子结[28]等超分子互锁体系,如图1所示。随着进一步细化,诸如[1]轮烷、[1]索烃等分子由两个或多个互锁组分通过共价键连接为一个共价分子的结构,又称为机械自锁结构(mechanically selflocked molecules)[29]。大部分超分子互锁体系都是先由主体和客体进行分子识别组装为准轮烷(一个动态的络合与解络合并存体系),再通过反应共价锁定体系而成。
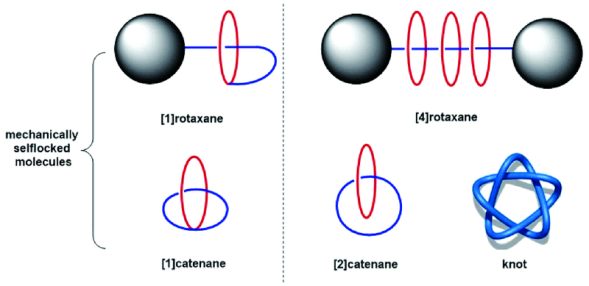
柱芳烃作为主体在构筑机械互锁结构方面取得了快速发展,自2011年Stoddart等[30]以7%的产率合成首例柱[5]芳烃[2]轮烷以来,经过近七年的发展,相继出现基于柱芳烃的[1]轮烷、[1]索烃、[2~5]轮烷、[2]索烃、雏菊链结构、轮烷树枝状分子、聚轮烷等,合成方法多样,产率也相应提高甚至达到定量合成。本文对近年来基于柱芳烃的超分子互锁结构的构筑、动力学研究及其功能化等领域进行阐述。
2 基于柱芳烃机械互锁结构的合成
2.1 基于柱芳烃的轮烷的合成
2.1.1 基于柱[5]芳烃的轮烷的合成
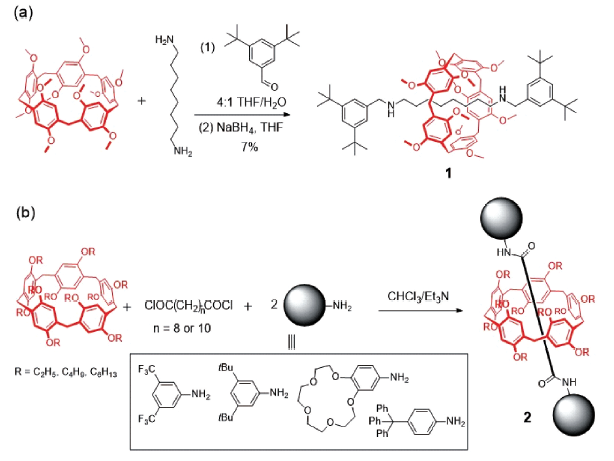
轮烷(rotaxane)是一类大环穿入线性分子并用封端基团将线性分子两端封闭锁定,使环不能脱落的互锁型分子。制备轮烷的方法有穿线封端法(Capping和Snapping)、滑动塞入法(Slipping)和钳夹合并法(Clipping)。2011年,Stoddart等[30]通过封端法利用1,4-二甲氧基柱[5]芳烃与1,8-辛二胺的识别作用,将伯胺基团与3,5-二叔丁基苯甲醛反应再用NaBH4还原,以7%的产率首次制备了柱芳烃[2]轮烷1(如图2a所示)。2012年,Huang等[31]在轴线上引入咪唑单元,增加主客体的络合作用(C—H…N和C—H…π氢键作用),以3,5-二硝基苯甲酸为封端基团,通过酰化酯化反应使合成轮烷的产率提高到23%。2015年,Nierengarten等[32]利用二酰氯衍生物与伯胺形成酰胺的封端反应制备[2]轮烷2(图2b),产率最高可达63%;他们还探讨了封端基团苯胺上取代基的电负性、二酰氯轴线烷基链长度以及柱[5]芳烃取代烷基长度对[2]轮烷产率的影响,为后续设计合成功能化[2]轮烷提供了参考。
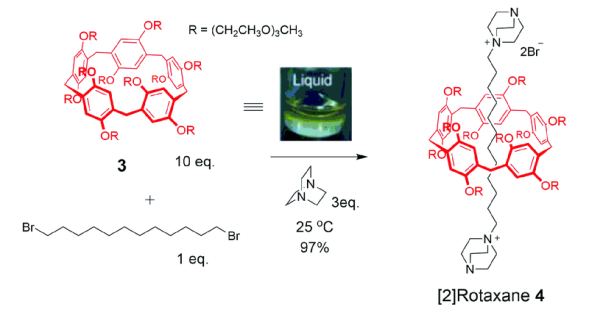
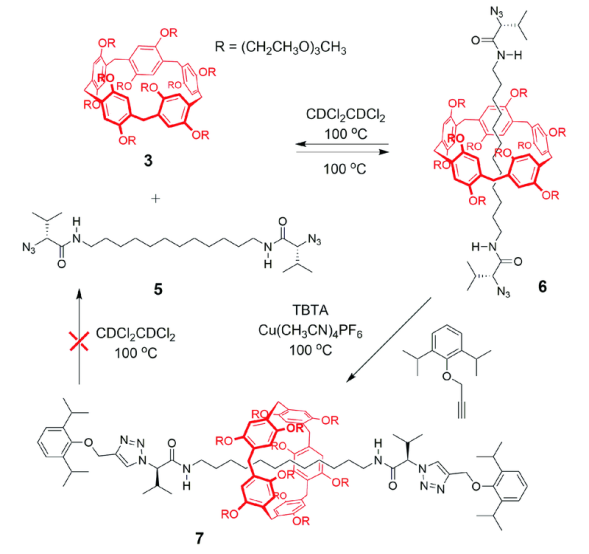
将两组分以1∶10的比例混合加热,在70 ℃下搅拌200 h,得到[2]轮烷6,产率约51%。该互锁结构在常温下稳定,当在CDCl2CDCl2溶液中加热至100 ℃,搅拌20 h以后,[2]轮烷6发生完全解离,得到柱[5]芳烃3和轴线分子5,这是因为溶剂分子的介入加速了解离,同时阻碍了主客体络合互穿的过程。进一步将[2]轮烷6进行“Click”封端反应,可得到高温下依然稳定存在的[2]轮烷7,主要因为封端基团增大阻止了高温下环从轴线上解离的过程。
钳夹合并法(Clipping)制备轮烷要求在成环过程中加入哑铃分子作为模板,通过哑铃分子与成环基元之间的非共价键作用,将各前体分子预组织在哑铃分子周围,再反应成环。一般来说,此法需要哑铃分子与成环前体分子之间有很强的非共价键作用力,且具有相应的空间匹配度。到目前为止,利用Clipping法制备柱芳烃轮烷的例子还未见报道。
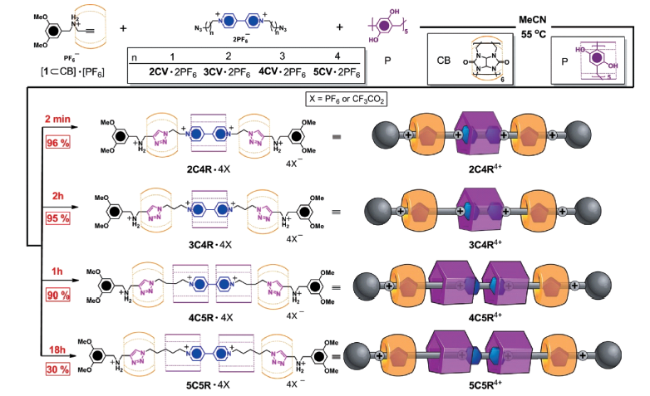
随着结构中独立组分增多,例如从[2]轮烷到[3]轮烷,其合成难度也相应增加。2012年,Ogoshi等[34]利用“Click”反应,在二乙氧基柱[5]芳烃存在下, 将1,3-二叠氮丙烷与含反式偶氮苯的炔烃衍生物连接,以45%的产率合成含两环一轴的[3]轮烷结构。2013年,Stoddart等[37]利用正交模板作用(orthogonal templation),通过葫芦脲[6]与全羟基柱[5]芳烃之间多氢键预组织作用,高效制备了[4]轮烷和[5]轮烷。如图5所示,当葫芦脲[6]铵盐复合物与联吡啶盐2CV混合时,以30%的产率生成[3]轮烷,且反应在55 ℃条件下进行2 d依然不完全;而加入全羟基柱[5]芳烃P后,55 ℃条件下2 min即可反应完全,且[4]轮烷的合成产率高达96%,这主要是因为CB[6]…P…CB[6]之间形成了氢键作用,将三个准轮烷体系预组织有效促进了铜催化叠氮端炔环加成(CuAAC)的反应;当含联吡啶的端炔链由2CV延长至3CV时,由于距离增大使CB[6]、P、CB[6]之间先产生CB[6]…P氢键作用,再产生CB[6]…P…CB[6]氢键作用,反应时间延长至2 h,[4]轮烷产率基本不变(95%);当端炔链由2CV延长至4CV时,距离的突增使CB[6]、P、CB[6]之间不能形成稳定的氢键作用,而生成了CB[6]…P…P…CB[6]的四组分氢键预组织体系,反应1 h,可以90%的产率得到[5]轮烷;增长端炔链为5CV时,四组分氢键作用减弱,反应时间延长至18 h,生成[5]轮烷的产率降至30%;端炔链为6CV时,反应一周时间只得到微量的[5]轮烷,表明氢键预组织作用因距离原因被破坏。此外,CB[6]与P之间的氢键作用对柱[5]芳烃P的构象翻转也有一定的影响,对于[5]轮烷4C5R,CB[6]…P…P…CB[6]为达到最优化的分子间氢键作用,使得两个P分子以A/A’的优势构象存在。
2.1.2 基于柱[6]芳烃的轮烷的合成
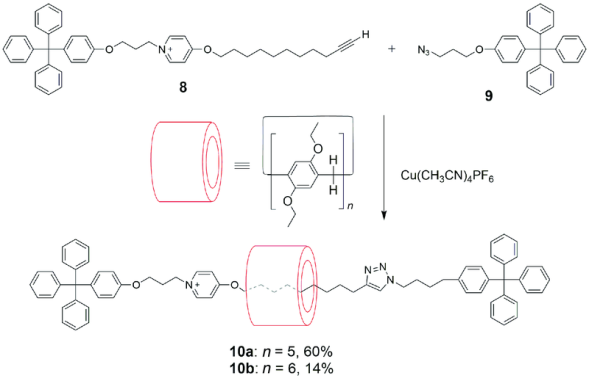
2012年,Ogoshi等[38]通过Snapping方法利用“Click”反应首次制备了基于柱[6]芳烃的[2]轮烷10b(图6)。由于柱[6]芳烃对吡啶盐的识别作用比柱[5]芳烃弱,[2]轮烷10b的合成产率(14%)低于相应柱[5]芳烃[2]轮烷10a的合成产率(60%)。Stoddart等[39]利用葫芦脲(CB)的协同作用,提高了柱[6]芳烃轮烷的产率。方法与图5类似,以端基为叠氮烷烃的二吡啶盐为轴,以含炔烃的葫芦脲-铵盐复合物做封端基团,在全羟基柱[6]芳烃存在下,利用叠氮-炔环加成反应以及葫芦脲与柱[6]芳烃之间的多氢键作用,较高产率(38%~68%)制备了基于柱[6]芳烃的[4]轮烷。
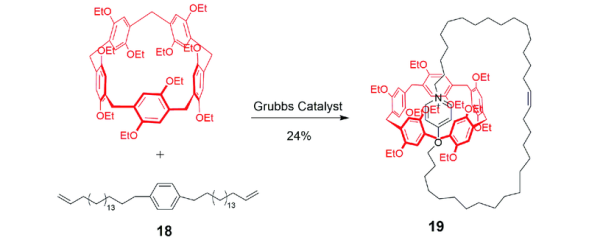
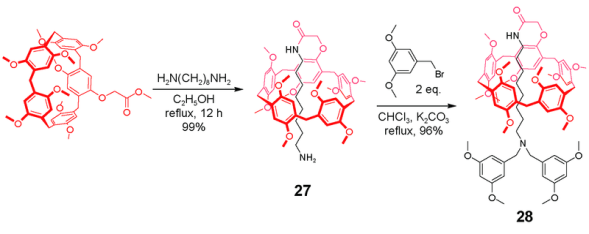
2.1.3 基于柱芳烃的雏菊链互锁结构的轮烷的合成
2.2 基于柱芳烃的索烃的合成
索烃[42]在超分子化学领域是一类重要的机械互锁结构,由于环与环之间互相锁定,在增加拓扑美学结构的同时,合成难度也相应提高。2013年,Huang等[43]报道了基于柱[5]芳烃的动态[1]索烃的合成和可逆调控。如图9所示,以含两个18-冠-6的柱[5]芳烃衍生物为主体,先与1,8-辛二胺形成[2]准轮烷,再加入三氟乙酸使客体酸化为铵盐,利用18-冠-6与铵盐的识别作用,形成动态[1]索烃(dynamic [1]catenane)结构,其中,基于柱[5]芳烃和18-冠-6两主体的识别作用互不干扰(orthogonal recognitions)。随后,加入过量三乙胺去质子化使得[1]索烃结构解离为动态[2]准轮烷结构,如此可实现pH调控的可逆过程。
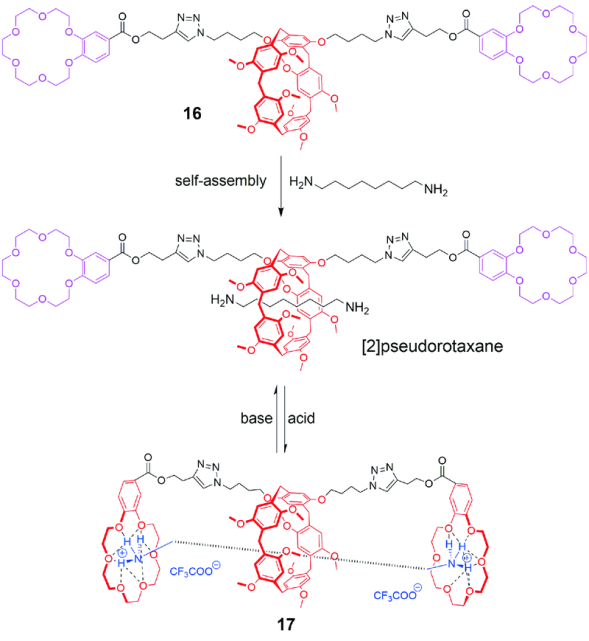
2.3 基于柱芳烃的机械自锁结构的合成
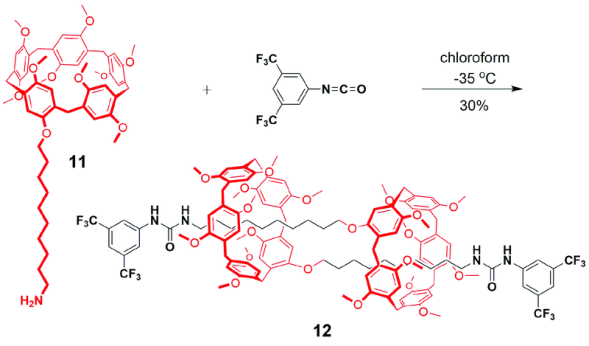
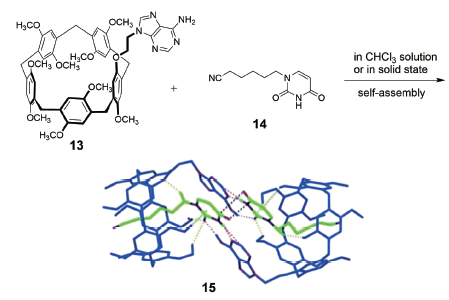
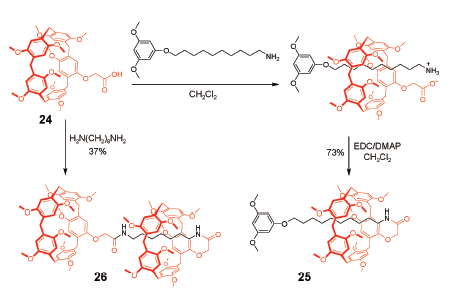
机械自锁结构[29]是一类结构特殊的超分子体系,是指单一组分通过非共价键作用自组装穿插锁定的分子结构。因为锁定的结构是单一组分,故称为自锁,所得结构包含[1]轮烷、[1]索烃、[1]准轮烷或[1]准索烃等。Ogoshi等[45]利用“Click”反应将1, 12-二叠氮十二烷与A1/A2二炔丙基柱[5]芳烃链接,以17%的产率生成完全互锁的[1]准索烃。该索烃可通过加入竞争客体或者不同极性的溶剂而构象翻转形成环外成环产物,这是由于“Click”反应的可逆性造成的。采取相似策略,强据莉等[46]利用异氰酸酯和伯胺反应成脲基衍生物的方法制备了具有溶剂极性和客体竞争调控性能的[1]准索烃。近期,Yang等[47]报道了一类柱[5]芳烃并甘醇链或烷基链成环的二环体系,温度或溶剂分子可逆调控环与环之间的内外翻转,该结构也形象地称为分子万向节(molecular universal joint),他们利用单晶衍射和圆二色谱等测试方法首次确定了[1]索烃结构的绝对构型。
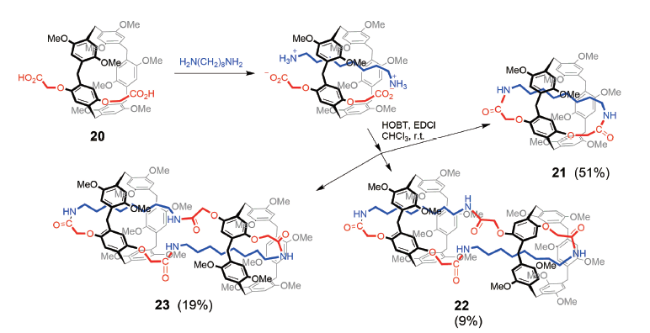
Liu等[29]利用1-羟基苯并三氮唑(HOBT)催化的酰胺反应制备了另一类结构稳定而不可逆的[1]准索烃和双体索烃。如图11所示,A1/A2二羧酸取代的柱[5]芳烃20与辛二胺在1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)和HOBT的催化下反应,可以生成自锁体系21~23,产率分别为51%、9%和19%。其中由于两个柱[5]芳烃单元构象不同,导致双体索烃22为对称结构,而23则为外消旋对映体结构。当使用丙二胺、丁二胺或己二胺时,[1]准索烃的合成产率提高,而当碳原子个数延长为8、10或12时,[1]准索烃产率降低,同时双分子互锁结构产生。这类双体索烃在机械互锁结构中较少见,与[1]准索烃同属分子自锁结构。
3 基于柱芳烃的机械互锁结构的分子运动
3.1 柱[5]芳烃轮烷的穿梭运动
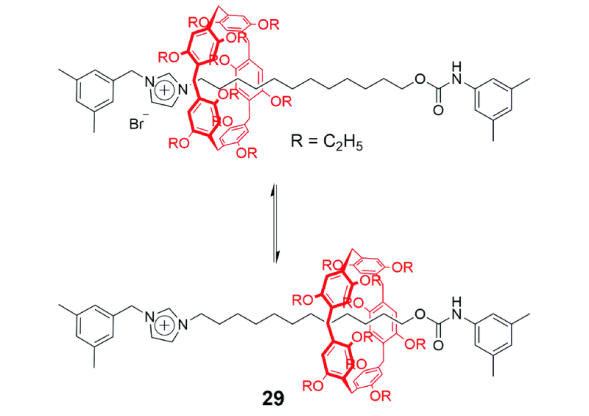
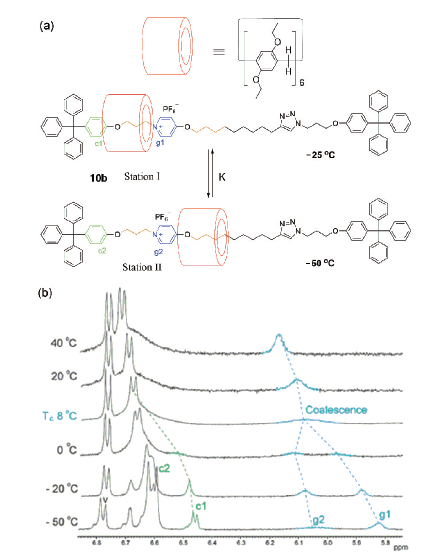
机械互锁结构由两个或以上独立组分互锁组成,研究各组分之间的相对运动有助于理解这类体系的分子动力学,也为构筑分子器件如分子弹簧、分子电梯等提供了基础。轮烷中大环延轴的穿梭运动是机械互锁结构中常见的互锁组分间的相对运动。在基于柱[5]芳烃的[2]轮烷29中(图14)[52],轴线一端含有咪唑盐,构成柱[5]芳烃的识别位点,另一端为单纯的烷基链,与柱[5]芳烃也存在较弱的C―H…π作用。在非极性溶剂氘代氯仿中,柱[5]芳烃环位于络合作用较强的咪唑盐位点,加入极性溶剂氘代二甲基亚砜(DMSO)时,由于DMSO破坏柱[5]芳烃和咪唑盐之间的氢键作用以及烷基链端的疏溶剂作用,使得大环远离咪唑盐移向烷基链端;研究还发现,在氘代DMSO中,常温加热至115 ℃时,柱[5]芳烃环又可从烷基链端移向咪唑盐位点,这可能是由两种识别作用对温度的依赖性不同导致的。总而言之,这类轮烷结构可利用溶剂的极性或温度来可逆调控大环在轴线的穿梭运动。类似的例子还有含硼二吡咯亚甲基染料(Bodipy)封端基团的[2]轮烷[53],柱[5]芳烃环通过溶剂极性和温度的调控做穿梭运动。随后,Liu等[54]利用自由能计算法对溶剂在[2]轮烷29穿梭运动中的影响做了详细探讨,认为溶剂的极性以及氢键键合能力共同影响了环在[2]轮烷中的位置。在氢键键合能力强的溶剂中,溶剂分子倾向于和轴链上的咪唑盐阳离子形成氢键作用,使得环移向烷基链部分,即使对极性较弱的乙醚溶剂来说,也同样如此。
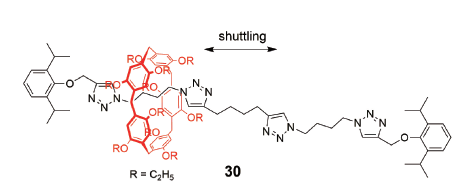
当轴线上两个环识别位点相同时,[2]轮烷30的穿梭运动因为溶剂的不同而结果不同(图15)[55]。在该[2]轮烷中,轴线上含四个1,2,3-三氮唑环,由于C―H…N氢键作用,柱[5]芳烃倾向位于与氮原子相接的亚丁基链段,而不在中间位置与碳原子相接的亚丁基链段。实验以及随后的自由能计算理论[56]详细探讨了溶剂对环在该[2]轮烷中的穿梭运动的影响,发现在DMSO中,环在轴线上的穿梭运动比在氯仿中速度更快,原因主要是DMSO的极性和与三氮唑环的氢键作用,降低了环运动的能垒。自由能计算理论还针对两个三氮唑环之间的烷基链长度对环穿梭运动的影响进行研究,分别采取碳原子个数为3~6的链段进行计算模拟,发现环的穿梭速率随着碳原子个数增加逐渐减慢,在碳原子个数为5时速度最慢;然而,近期的实验结果却与此结果相悖。Ogoshi等[57]制备了一系列中间以不同长度的碳链或甘醇链连接的两端分别含三氮唑丁基的[2]轮烷,通过变温核磁实验发现,当两个识别位点连接单元都为碳链或者甘醇链时,环穿梭运动的活化自由能与连接单元的长度无关,这是因为活化自由能大小是由环离开三氮唑进入连接单元的能垒决定的,环在碳链(或甘醇链)上的穿梭运动自由能较低;但是连接单元的化学结构会影响自由能大小,当烷基链换为甘醇链时,由于氧原子与负电空腔环的排斥作用,使得环往返运动的活化自由能增大。研究加深了对环在轮烷上穿梭运动的动力学认识,为进一步设计基于柱芳烃的分子机器奠定了基础。
3.2 柱[6]芳烃轮烷的穿梭运动
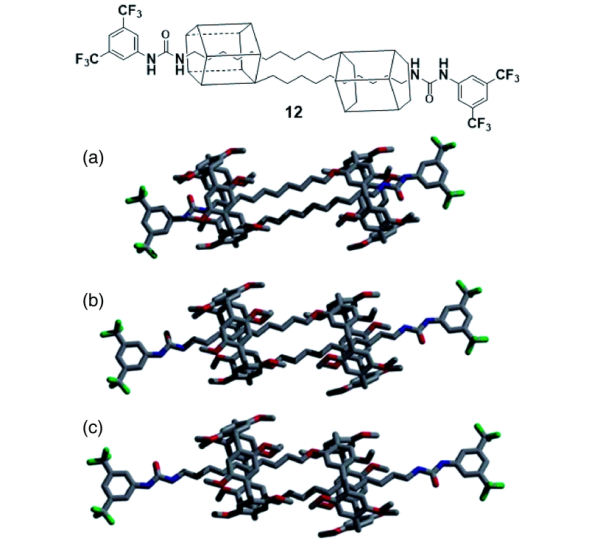
3.3 雏菊链互锁结构的伸缩运动
在雏菊链式互锁交叉结构中,环在链间的穿梭运动更为复杂[40]。如图17a所示,在氘代氯仿中,雏菊链12以收缩的优势构象存在,可能是两个烷基链间的色散力导致的,此时该超分子结构长度为31 Å;在氘代DMSO中,轮烷互锁二聚体12以拉伸的优势构象存在(图17c),可能是因为极性的DMSO分子与烷基链间更强的诱导力破坏了色散力的平衡,此时该超分子结构长度为37 Å;当往氘代氯仿溶液中滴加氘代DMSO时,随着极性增加,该互锁结构由收缩状态逐渐持续拉伸延长,就像弹簧的拉伸运动,例如在氯仿与DMSO体积比为2∶1时,分子长度为35 Å(图17b),该体系被形象地称作“分子弹簧”。这类持续的伸缩运动在超分子体系中较为少见,为模拟人体肌肉的伸缩运动奠定了基础。
4 基于柱芳烃的机械互锁结构的功能化
4.1 柱芳烃机械互锁结构的手性翻转
柱芳烃由于衍生基团的弹性翻转而构象多样化,含较小取代基的柱[5]芳烃一般存在互为镜面对称的8种异构体(图18a所示),分别为:(pS, pS, pS, pS, pS)、(pR, pR, pR, pR, pR)、(pR, pS, pS, pS, pS)、(pS, pR, pR, pR, pR)、(pR, pR, pS, pS, pS)、(pS, pS, pR, pR, pR)、(pR, pS, pR, pS, pS)和(pS, pR, pS, pR, pR)。固定构象的方法可以是引入较大的取代基限制翻转,也可以是加入其他组分形成机械互锁结构,通过其他组分的介入限制柱芳烃取代基的翻转。例如,在[2]轮烷31和[3]轮烷32[34]中(图18b),由于轴线分子的穿入,只有能量较低、环空腔较大的镜面对称构象(pS, pS, pS, pS, pS)和(pR, pR, pR, pR, pR)大环才能形成轮烷,其他几个异构体均不能形成轮烷结构。但是,对于全羟基柱[6]芳烃的[4]轮烷33[39]而言,由于环空腔较大,衍生基团较小,在轮烷中依然存在多种异构体,只是有优势倾向的选择。在双体索烃中(图11)[29],由于柱[5]芳烃的镜面对称出现了中心对称非手性的索烃22和无对称中心的手性外消旋对映体索烃23。
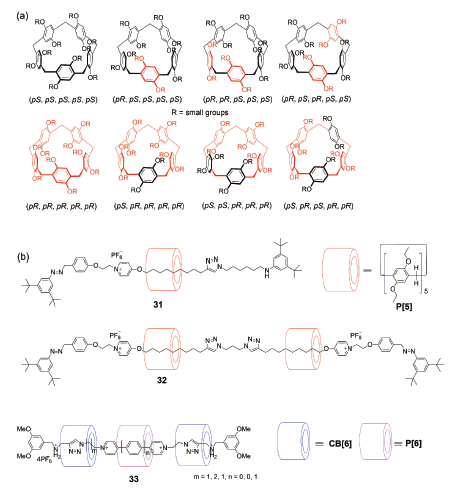
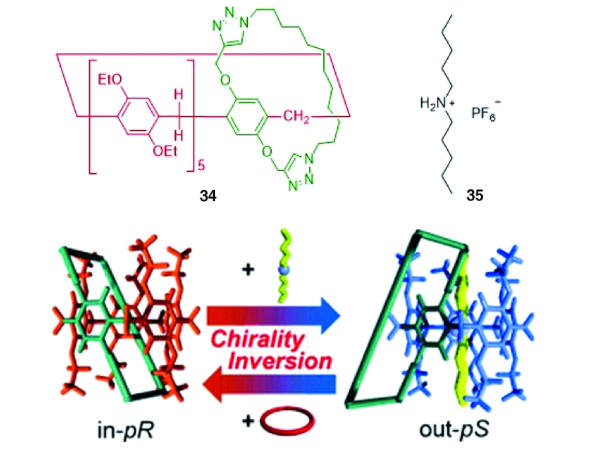
除了溶剂和新的客体分子可导致索烃手性翻转外,Yang等[47]发现在分子万向节中,温度也可以调节该体系的手性,这在超分子体系中颇为少见。这类分子的绝对构型可随温度降低由in-Sp转为out-Rp,转变程度可由各向异性因数g(anisotropy factor)的变化表示,Δg的大小和溶剂分子有关,同时也受到甘醇链或碳链长度的影响,当该结构中甘醇链个数为4时,在甲基环己烷溶剂中,温度从10 ℃降到-90 ℃可使Δg高达0.03,这是目前为止所有报道中Δg的最大值。
4.2 柱芳烃机械互锁结构的荧光能量转移
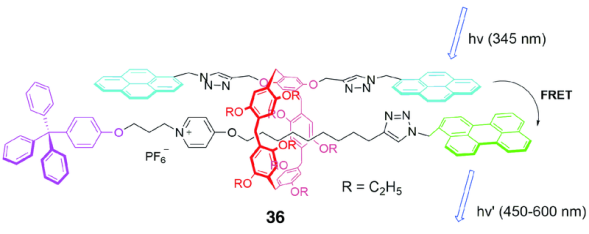
荧光共振能量转移(Förster resonance energy transfer,FRET)[58]是当下生命科学领域的研究热点之一,在研究生物大分子之间的相互作用、细胞生理研究等方面有重要的应用价值。两个荧光基团要实现FRET需满足一定的条件,供体的发光光谱与受体的激发光谱要重叠,同时两者距离很近,位置合适。机械互锁结构为实现分子与分子之间的FRET提供了基础,对于图20所示的[2]轮烷36[59],两个芘基团位于柱[5]芳烃的A1/A2两侧位点,一个苝基团位于轴链的一端,因为轮烷的互锁结构使芘和苝的距离非常接近。紫外吸收和荧光光谱发现,芘衍生柱[5]芳烃的激发波长与单独的含苝哑铃分子的吸收光谱有部分重叠,使得[2]轮烷在345 nm光谱激发时,发生FRET现象,出现高强度的激发光谱,这在环与客体分子单独存在的稀溶液中是没有发生的。
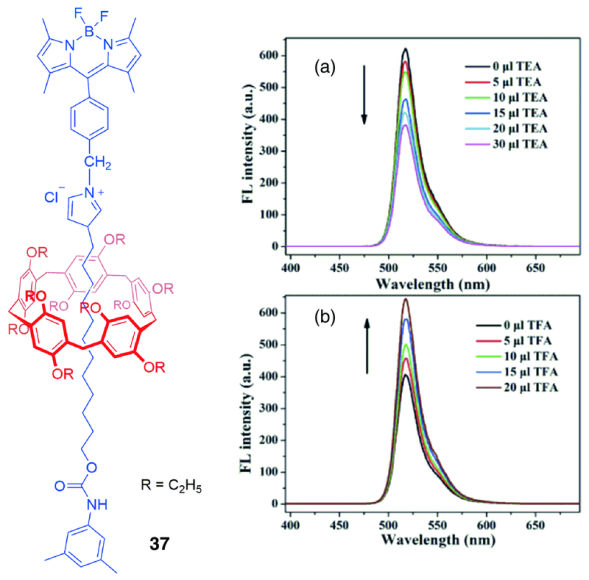
4.3 柱芳烃机械互锁结构的响应性凝胶
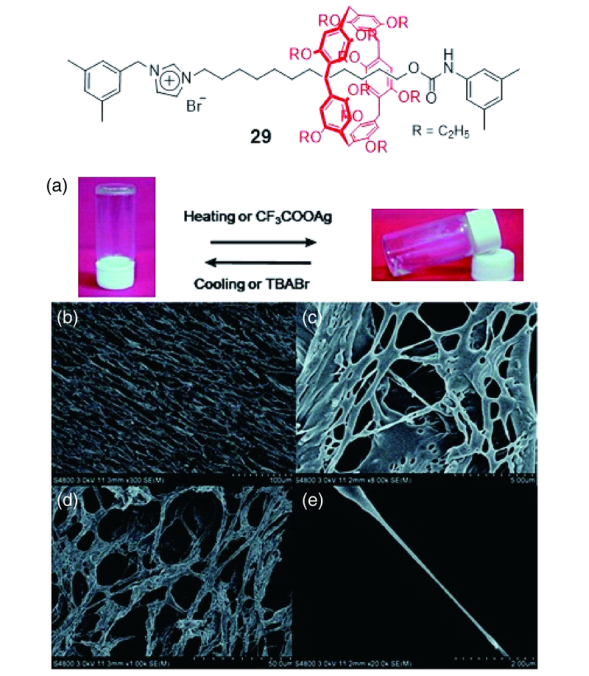
柱芳烃[2]轮烷29(图14)[52]的咪唑溴盐在DMSO溶剂中可以看作一种两亲分子(咪唑盐端亲溶剂,而烷基链端疏溶剂),这种两亲性使分子自组装为纳米纤维(直径约200 nm,长度几百微米),纳米纤维之间进一步交联为网状结构,将溶剂分子包裹在其内部,形成凝胶,图22b~e是该凝胶冷冻干燥后在扫描电镜(SEM)下的纤维网格状图像。这类凝胶还具备一定的响应性(图22a),加入CF3COOAg后发现,凝胶结构破坏变为澄清溶液,原因可能是阴离子CF3COO-和Br-之间发生交换,破坏了分子原有的自组装行为,再加入四丁基溴化铵(TBABr)时,咪唑盐又与溴离子配对,恢复原有的自组装结构,凝胶再次生成;此外,升温可使凝胶转变为溶液,降温又使溶液变为凝胶。这种对温度和化学试剂的响应性,有望扩大此类超分子凝胶的潜在应用。
4.4 柱芳烃机械互锁结构的Langmuir膜
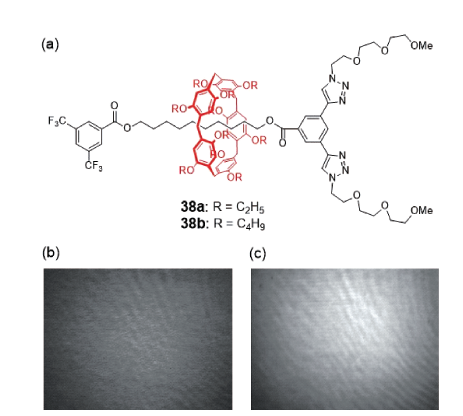
Langmuir膜[60]是一类两亲性的分子级纳米膜,由于其超大的表面积而具有特殊的物理化学性质。Nierengarten等[61]利用疏水的烷基链柱[5]芳烃(二乙氧基柱[5]芳烃和二丁氧基柱[5]芳烃)和含亲水基甘醇链的封端基团,在烷基链轴线分子穿线作用下制备了两亲性[2]轮烷38a,b(图23)。研究发现,[2]轮烷的两亲性达到一定的平衡,使得分子在水相-空气的界面上形成Langmuir膜,图23b和图23c分别为[2]轮烷38a在表面压力45 mN/m作用下和[2]轮烷38b在表面压力12 mN/m作用下所形成的Langmuir膜;柱[5]芳烃环上的烷基链长度对于膜在压缩-解压缩过程中是否可逆起到关键作用,同时环在轴链上的穿梭运动对膜中分子间的堆积和排列也有一定的影响。
4.5 柱芳烃轮烷树枝状分子的构筑和功能化
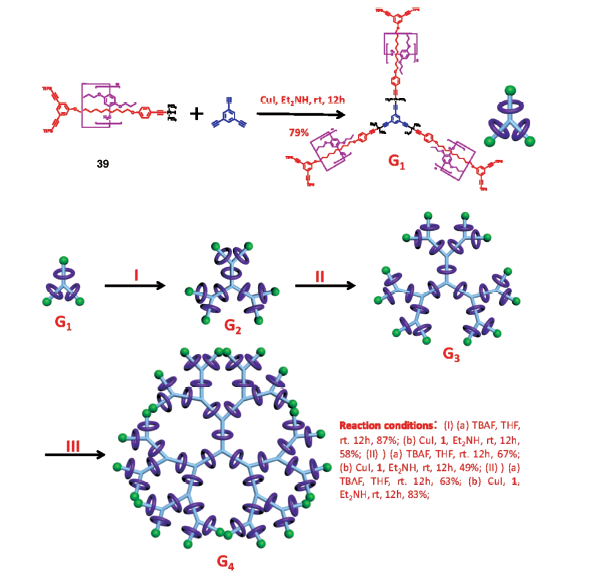
树枝状分子是一类具有多个重复单元的树枝形发散的独特分子类型[62]。将轮烷结构引入到树枝状分子中,使得分子同时具有高支化和分子内相对运动的特点,有望赋予分子新的功能。2015年,Yang等[63]首先制备了有机金属[2]轮烷39(图24),可以稳定地溶解于多种常见溶剂;以此为重复单元,与1,3,5-三乙炔苯进行Cu(Ⅰ)催化偶联反应,得到第一代轮烷树枝状分子G1,产率为79%;进而将G1端基炔基去保护再与[2]轮烷39偶联反应,生成第二代轮烷树枝状分子G2;用类似的方法将低代数轮烷树枝状分子依次进行脱保护-偶联反应,可得到高代数轮烷树枝状分子G3和G4,其中G4分子中含有45个[2]轮烷单元。与不含柱[5]芳烃的模型树枝状分子相比,柱[5]芳烃环的存在增加了支链的刚性,阻止了支链的自缠绕并减少了支链断裂现象。进一步研究表明,这类含多炔烃的轮烷树枝状分子可以通过表面化学转化法引入多个二茂铁进行功能化。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
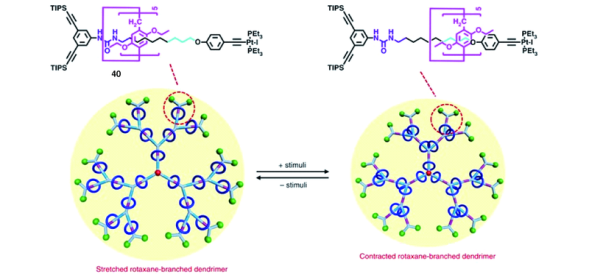
环与轴链分子间的相对运动是轮烷最重要的特征之一,为了在轮烷树枝状分子中实现多环的相对运动,Yang等[64]改进了上述轮烷树枝状分子结构(图25),在[2]轮烷40轴线上引入不同于烷基链的脲基识别位点。首先合成[2]轮烷,再采取图24的方法制备了新型G1~G3代轮烷树枝状分子。由于环与脲基间具有比C―H…π更强的多氢键作用,使得[2]轮烷在溶剂四氢呋喃中处于脲基位点处;加入氢键竞争溶剂DMSO或者四正丁基乙酸铵试剂(TBAA),可使[2]轮烷大环远离脲基移向烷基链另一端。同样,在轮烷树枝状分子G1~G3中也可以实现加入DMSO或TBAA后多环的运动(图25)。有趣的是,当环位于脲基位点时,增加了各支状链段靠近外沿处的刚性,分子处于伸展状态,尺寸较大;当环位于另一端烷基链时,靠近外沿的支链端刚性减弱,链段间更容易进行自缠绕,因而树枝状分子尺寸减小。由此实现了尺寸可调控的轮烷树枝状分子,为进一步实现超分子体系的分子捕获、传递或催化等功能提供了条件。
5 结论与展望
基于柱[5]芳烃的轮烷、索烃等机械互锁结构合成方法多样,产率最高近乎定量,体系种类丰富;以柱[6]芳烃为主体构筑的互锁体系相对单一,而柱[7~15]芳烃由于合成产率低,超分子互锁体系的构筑难以实现。伴随着柱芳烃合成方法的突破,基于高阶柱芳烃构筑机械互锁结构的前景有待实现。
柱芳烃机械互锁结构动力学研究主要集中于柱芳烃在轮烷轴线上的穿梭运动,对于环在轮烷轴线或者索烃环中的旋转运动却未见报道,为今后发展方向之一;此外,环在互锁结构中的相对运动已逐渐由单分子运动向多分子同时运动的趋势发展,例如柱[5]芳烃轮烷树枝状分子中的多环运动,有望构筑更复杂的甚至宏观可见的分子器件。
柱芳烃机械互锁结构可实现多功能化,例如手性翻转、荧光能量共振转移、响应性凝胶、Langmuir膜等,随着柱芳烃更为广泛的衍生化,基于柱芳烃的超分子互锁体系将更加多样化,必将具备更多功能。例如近期研究发现,含叔胺的[1]轮烷可以作为催化剂成功催化Knoevenagel缩合反应[51],而更广阔未知的应用领域也将随着研究的深入逐步展开,成为柱芳烃机械互锁结构更具挑战性的发展前景。