



Contents
1 Introduction
2 Two-coordinated 3d-SIMs
3 Three-coordinated 3d-SIMs
4 Four-coordinated 3d-SIMs
5 Five-coordinated 3d-SIMs
6 Six-coordinated 3d-SIMs
7 High-coordinated(seven and eight) 3d-SIMs
8 Theoretical calculation
9 Conclusion and outlook
1 引言
当3d过渡金属离子具有T基谱项时,一级轨道角动量未被猝灭,产生显著的自旋-轨道耦合,例如低自旋的d4(MnIII, CrII)和d5(FeIII)、高自旋的d6(FeII)和d7(CoⅡ)等。首例3d过渡金属单离子磁体(3d-SIM)就是一例高自旋FeII的单核化合物K[(tpaMes)Fe][6]。自此之后,3d-SIMs日益增多。而这种单离子磁体结构更简单,易于研究磁构关系,也为开发新型单分子磁体提供了一个新的思路。本文主要总结了3d-SIMs的研究进展。
2 二配位的3d-SIMs
二配位是配合物中最低的配位数,而这种线性二配位的结构可产生非常大的单轴磁各向异性,从而获得较高的翻转能垒。但是此类化合物一般不稳定。例如,2013年,Zadrozny等报道了一系列二配位Fe(Ⅱ)的单离子磁体[13]。他们合成了6个不同的二配位Fe(Ⅱ)的化合物,分别为:Fe[N(SiMe3)(Dipp)]2(Dipp=C6H3-2,6-Pr2i)、Fe[C(SiMe3)3]2、 Fe[N(H)Ar’]2(Ar’=C6H3-2,6-(C6H3-2,6-Pr2i)2)、 Fe[N(H)Ar*]2(Ar*=C6H3-2,6-(C6H2-2,4,6-Pr3i)2)、 Fe(OAr’)2和Fe[N(H)Ar#]2 (Ar#=C6H3-2,6-(C6H2-2,4,6-Me3)2)。前5个化合物的L—Fe—L键角都等于180°,配位构型为直线,而最后一个化合物的L—Fe—L键角为140.9(2)°,呈V字构型。理论计算发现L—Fe—L键角严重影响d轨道的能级分布。当L—Fe—L键为线性时,dxy和 轨道接近简并,而V字构型的L—Fe—L键角导致dxy和 轨道能量相差很大,这将可能降低磁各向异性。最后一个化合物的虚部交流磁化率只表现出频率依赖,并没有得到相应的自旋翻转有效能垒Ueff,而前五个化合物的Ueff值分别为181、146、109、104和43 cm-1。
近年来,高松课题组基于二配位构型在钴基单离子磁体上取得了重要的突破。他们于2017年报道了一系列基于亚胺配体的二配位的单核Co(Ⅱ)配合物。这种亚胺中的N与Co(Ⅱ)形成了较短的Co=N双键(1.691(6)Å),从而增强了这种直线型配合物的磁各向异性。磁学研究表明,该系列配合物在零场下表现出单离子磁体性质,其中一例配合物的能垒达到了413 cm-1,是目前含过渡金属的单分子磁体中最高的有效能垒[15]。
众所周知,量子隧穿效应的存在会湮灭或减弱单分子磁体性质。二配位构型易于形成轴向的直线型配体场,有利于获得性能优良的单离子磁体。但是,这种直线构型可能存在着动态的无序,从而使其很难保证轴对称,导致存在横向零场分裂能E,促使量子隧穿效应的产生。比如,Samuel等利用卡宾配体合成了一例二配位的一价铁的单离子磁体[(cAAC)2Fe][B(C6F5)4][16]。该化合物仅仅表现出场致单离子磁体性质,能垒低于20 cm-1。Werncke等研究了二配位的Fe(Ⅰ)基单离子磁体([Fe{N(SiMe3)2}2]-)的性能。由于该系列配合物的对称性较低,它们的能垒明显小于同类的单离子磁体[17]。高松课题组合成出了首例二配位的d8电子构型的Co(Ⅰ)基单离子磁体[18]。该配合物同样未表现出零场慢磁弛豫行为,能垒也仅达到21.3 cm-1。利用大空间位阻的N-杂环卡宾配体与Ni(Ⅰ)结合得到线性的二配位Ni(Ⅰ)化合物[Ni(6-Mes)2]Br[19]。有趣的是,Ni(Ⅰ)的自旋值 S =1/2,但是该化合物却表现出单离子磁体行为。这归因于在基态时轨道角动量并没有淬灭,从而导致一级自旋-轨道耦合,产生磁各向异性。
3 三配位的3d-SIMs
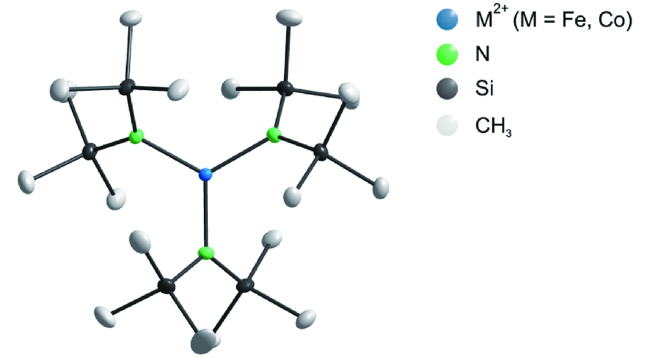
三配位配合物的构型大多数是平面三角形,这种构型往往不易于产生单轴磁各向异性,因此,此类单离子磁体的性能并不理想。Lin等报道了一例具有平面三角形构型的[FeII(N(TMS)2)2(PCy3)](TMS=SiMe3,Cy=cyclohexyl)。该配合物在外加600 Oe下表现出慢磁弛豫行为[20]。Eichhöfer等报道了三角平面构型的Fe(Ⅱ)的化合物[Fe{N(SiMe3)2}2(PCy3)]和Co(Ⅱ)配合物[Co{N(SiMe3)2}3]、[Co{N(Si-Me3)2}2(THF)]和[Co{N(SiMe3)2}2(PCy3)](图2)。这些化合物在加场的情况下出现单离子磁体性质[21]。CASSCF/SOCI理论计算表明体系中磁各向异性都来源于激发态与基态发生混合所产生的自旋-轨道耦合作用。在此基础上,他们利用类似的方法又合成了两例三配位的Ni(Ⅰ)基单离子磁体[NiCl(PPh3)2]·C4H8O和[Ni(N(SiMe3)2)(PPh3)2][22]。
郑彦臻课题组合成了一例具有平面三角配位构型的单核Co(Ⅱ)配合物[Li(THF)4][Co(NPh2)3],其构型接近于D3h对称性。直流磁化率数据和ERP测试表明该配合物具有较大的、正值的零场分裂能,而理论计算表明这种正值的D是由二阶自旋-轨道耦合产生的。交流磁化率表明该配合物具有场致慢磁弛豫性质[23]。他们利用2,6-二叔丁基苯酚配体又合成了两例平面三角型的配合物[Na(THF)6][Co(OAr)3] 和[(THF)3NaCo(OAr)3]。这两个配合物具有相同的配位数和配体,但是由于抗衡离子的不同导致配位构型的对称性发生改变,前者为C2v对称,而后者属于Cs对称。两个配合物都具有大且负的D值,其数值相近,但是前者配合物具有单离子磁体性质,而后者的交流磁化率并没有表现出频率依赖。理论计算表明,在后者配合物中,由于其相对较低的对称性导致了系统中存在较大的横向零场分裂能,从而产生量子隧穿,抑制了慢磁弛豫的发生[24]。
4 四配位的3d-SIMs
四配位构型主要有三角单锥和四面体。三角单锥构型的单离子磁体主要存在于Fe(Ⅱ)的配合物中,而在Co(Ⅱ)基单离子磁体中并没有出现。相对来说,四面体构型就比较常见。
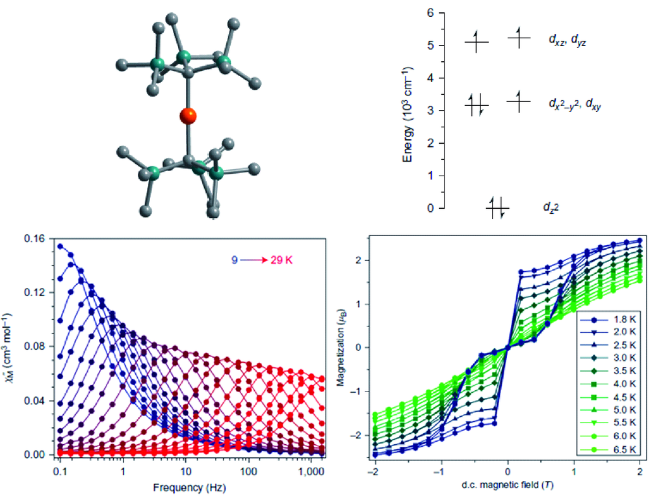
2010年,Long课题组等报道了一例三角单锥型的Fe(Ⅱ)基单离子磁体K[(tpaMes)Fe],如图3[6],这是第一例过渡金属单离子磁体。拟合磁化强度曲线得到该化合物的零场分裂能D=-39.6 cm-1。在交流磁化率测试中,该化合物在1500 Oe下出现慢磁弛豫行为,其能垒Ueff为42 cm-1。随后,他们通过改变配体得到了一系列类似的配合物[(tpaR)Fe]-(R=tert-butyl; phenyl; mesityl; 2,4,6-triisopropylphenyl; 2,6-difluorophenyl)[26]。直流磁性测量表明这5个化合物均具有单轴磁各向异性,零场分裂能D分别为-48、-44、-30、-26和-6.2 cm-1,证实了零场分裂能随着Fe金属中心的配位场强度增加而增加。交流磁化率表明前三个化合物均为单离子磁体,Ueff值分别为65、42和25 cm-1。

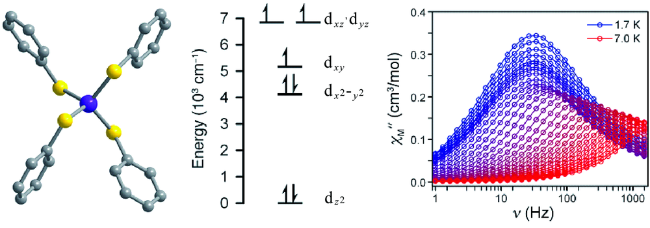
四面体可以说是Co(Ⅱ)基单离子磁体中最为经典的构型。Long课题组报道了首例钴基单离子磁体(Ph4P)2[Co(SPh)4][29],如图4所示。该配合物呈四面体构型。角重叠模型理论计算表明充满电子的 轨道与半满的dxy轨道能量较接近,导致激发态与基态发生混合,产生较强的自旋-轨道耦合。在未加场的情况下该化合物具有慢磁弛豫过程。通过合成(Ph4P)2[Co0.09Zn0.91(SPh)4]进行磁稀释研究表明这种量子隧穿效应是由分子间的偶极-偶极相互作用产生的。接着他们比较研究了含有不同配位原子O、S和Se的酚类配体配合物(Ph4P)2[Co(OPh)4](CH3CN)、K(Ph4P)[Co(OPh)4]、(Ph4P)2[Co(SPh)4]和(Ph4P)2[Co(SePh)4]的磁性质[30]。通过拟合磁化强度得到这4个化合物的零场分裂能分别为D=-11.1、-23.8、-62和-83 cm-1。研究表明,零场分裂能随着配位原子的软度增加而变大。交流磁化率测试表明这4个化合物均表现出单离子磁体行为。除了第1个化合物需要施加外场,其他3个配合物在零场下均能表现出慢磁弛豫行为。
Rechkemmer等发现四面体构型的单离子磁体(HNEt3)2[Co(L2-)2](H2L=1,2-bis(methanesulfon-amido)benzene)呈现出较高的有效能垒值,达到了118 cm-1,是四配位Co(Ⅱ)基单离子磁体中的最高能垒[31]。Buchholz等利用双齿配体合成了一系列稳定的Co(Ⅱ)基单离子磁体[Co(L2)](L1=2-(4,5-diphenyl-1H-imidazol-2-yl)phenol; L2=2-(4,5-diphenyl-1H-imidazol-2-yl)-4-nitrophenol; L3=2-(4,5-diphenyl-1H-imidazol-2-yl)-6-methoxy-phenol),其配位构型均为四面体[32]。施展课题组对三种具有扭曲四面体结构的化合物[Co(PPh3)2Cl2]、[Co(DPEphos)Cl2]和[Co(Xantphos)Cl2](PPh3=triphenylphosphine, DPEphos=2,2'-bis(diphenyl-phosphino)diphenyl ether, Xantphos=9,9-dimethyl-4,5-bis(diphenyl-phosphino)xanthenes)进行磁性研究发现,该三种化合物在外加磁场下均存在慢磁弛豫现象[33]。曹登科等报道了扭曲四面体构型的单核Co(Ⅱ)配合物Co(hpbdti)2·3CH3OH(hpbdtiH=2-(2-hydroxyphenyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole)的磁性质[34]。值得关注的是,该化合物在外加场下表现出多步慢磁弛豫行为,并且具有光致变色性质。该化合物在低温区表现出双弛豫过程,而在高于5.2 K时表现出单弛豫过程。这是因为该化合物存在分子间的π…π堆积相互作用和氢键,而在低温时这种微弱的相互作用比较明显,从而形成Co离子的二聚体,导致单轴磁各向异性非共线;然而,在高温时分子的热振动占主导,消除了分子间的相互作用。Bo$\breve{c}$a等也发现一例具有双弛豫过程的单离子磁体[Co(PPh3)2Br2][35]。在这个化合物中分子间并不存在相互作用,因此产生双弛豫过程的原因还有待于进一步的研究。近期,具有赝四面体结构的两例单核配合物Co(PNP)X2(X=Cl; X=SCN; PNP=bis(2-(diphenylphosphaneyl)-4-methylphenyl)amine)表现出单轴磁各向异性和场致单离子磁体性质[36]。
单轴磁各向异性(D < 0)一直以来是单分子磁体的关键条件,而易面磁各向异性(D > 0)似乎难以获得单分子磁体性质。然而,Long课题组在2012年首次发现了一例四配位的单核Co(Ⅱ)化合物[(3G)CoCl](CF3SO3)(3G=1,1,1-tris-[2 N-(1,1,3,3-tetramethylguanidino)ethyl]ethane)在零场分裂能为正值(D> 0) 的情况下也呈现出单离子磁体行为[37]。通过高场高频电子顺磁共振谱(HF-EPR)测试,得到该化合物的零场分裂能D为+12.7 cm-1。他们推测,由于声子瓶颈所导致的直接过程比较慢,从而产生慢弛豫现象。吴大雨等利用2, 9-二甲基-1, 10-菲啰啉(dmph)配体合成了四面体构型的单离子磁体[dmphCoBr2][38]。HF-EPR测试表明该化合物的D> 0。有趣的是,这种易面磁各向异性(D > 0)的现象同样存在于高自旋的Re(Ⅳ)-SIMs中(S=3/2)[39,40,41],而在其他过渡金属单离子磁体中并没有被发现。
具有平面正方形构型的单离子磁体极其少见。郑彦臻课题组报道了两例平面正方形的Cr(Ⅱ)基单离子磁体[Cr(N(TMS)2)2(L)2](TMS=SiMe3, L=pyridine 和tetrahydrofuran)。HF-EPR测试表明这两例配合物均具有单轴磁各向异性[42]。
利用环戊二烯基配体可以构筑出一类特殊四配位的单核配合物。环戊二烯环以π键与金属离子在一侧结合,另一端金属离子与芳香环提供的C原子进行配位,这种配位构型虽属于四配位,但是构筑了类直线型的配体场,有利于提高磁各向异性。Weismann等报道了一例金属有机Fe(Ⅱ)配合物[5CpFe(C6H3iPr2-2,6)](5Cp=C5iPr5)。该化合物中Fe(Ⅱ)与五异丙基环戊二烯基和2,6-二异丙基苯基上的一个C原子配位。拟合等温磁化曲线表明该化合物具有较大且负的零场分裂能D和相对较小的E值(E/D=0.006)。进一步的磁性测试表明,该化合物在外加直流场下表现出双弛豫行为。通过施加750和2500 Oe直流场可以分别得到两个独立的弛豫过程。在外加直流场为750 Oe时的弛豫过程的Ueff为40.3 K;而在外加直流场为2500 Oe时的弛豫过程的Ueff值为143.3 K[43]。而Chakraborty等获得了一例类似结构的Fe(Ⅰ)的单离子磁体[CpArFe(IiPr2Me2)](CpAr =C5(C6H4-4-Et)5; IiPr2Me2 =1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene)[44]。经穆斯堡尔谱、EPR谱、磁学数据以及理论计算表明该配合物具有S=3/2基态自旋和大的单轴磁各向异性(D < 0),其有效能垒Ueff为64 cm-1。
5 五配位的3d-SIMs
五配位的铁基单离子磁体目前仅有几例。2012年,Mossin等报道了空气稳定的含PNP钳合配体Fe(Ⅲ)配合物[(PNP)FeCl2](PNP=N[2-P(CHMe2)2-4-methylphenyl]2-)[45]。该化合物具有扭曲的三角双锥构型,同时具有单离子磁体和自旋交叉行为。磁性测量表明在80 K时呈现出S=3/2到S=5/2的自旋交叉转变。该化合物在零场下就能表现出慢磁弛豫行为,并获得它的有效能垒为32~36 cm-1。这是第一例三价铁基单离子磁体。2017年,Nocera课题组构筑了两例五配位的三价Fe单离子磁体:(PMe3)2FeCl3和(PMe2Ph)2FeCl3[46]。测试和计算表明前者的零场分裂能D=-50 cm-1,而后者的零场分裂能D=-17 cm-1。此外,这两个配合物的Ueff值分别为81和46 cm-1。后者的对称性明显低于前者。这也表明提高配合物的对称性可以在一定程度上提高单离子磁体的有效能垒。
一种三角双锥构型的单核Ni(Ⅱ)配合物[Ni(MDABCO)2Cl3]ClO4被报道具有巨大的磁各向异性。交流磁化率测试表明该配合物表现出慢磁弛豫行为,归属于拉曼和直接共同作用的自旋-晶格弛豫过程[47]。
2011年,Jurca等报道了首例五配位的Co(Ⅱ)基单离子磁体[48]。他们通过双(亚氨基)吡啶螯合配体得到扭曲的四方锥的配合物[{ArNdCMe}2(NPh)]Co(NCS)2和[{ArNdCPh}2(NPh)] Co(NCS)2。前者中的Co离子比4个N原子所形成的平面高0.39 Å,后者中为0.52 Å。理论计算表明,正是由于两个配合物中的Co离子与N平面之间存在距离,从而导致d轨道能级分裂产生自旋-轨道耦合作用。如图5所示,通过磁性测量发现这两例配合物均具有场致慢磁弛豫行为。他们基于三联吡啶配体得到了两种五配位的不同构型的单核Co(Ⅱ)的配合物[Co(terpy)Cl2]和[Co(terpy)(NCS)2][49]。前者为四方锥构型,后者为扭曲的三角双锥。当施加外场时,两者都表现出单离子磁体行为,并且都出现双弛豫过程。通过施加两个场,可以得到这两个弛豫过程的有效能垒Ueff。在前者化合物中,快弛豫过程的Ueff值为28 K,慢弛豫过程的能垒为4 K; 在后者化合物中,快弛豫过程的Ueff值为17 K,慢弛豫过程的Ueff为3 K。
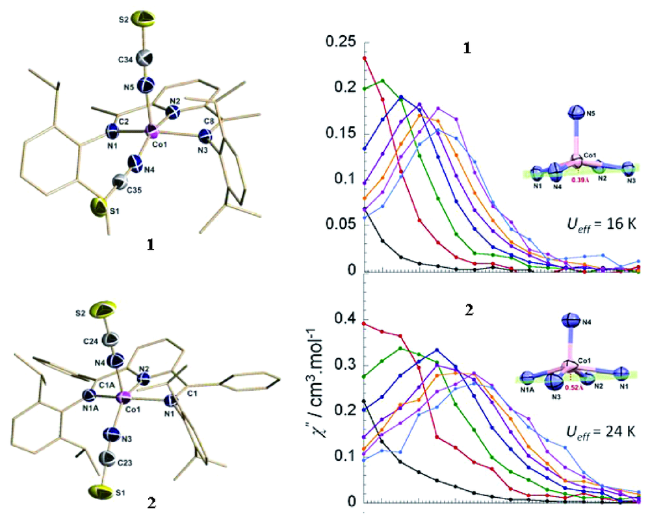
Ruamps等研究了两例具有三角双锥构型(C3v对称性)的单核Co(Ⅱ)配合物[Co(Me6tren)Cl]ClO4和[Co(Me6tren)Br]Br的磁性质[50]。磁性测量、HF-EPR测试和理论计算都表明两个化合物的零场分裂能D均为负值。研究认为赤道平面的配体提供弱σ作用和轴向的配体提供强π作用有利于形成较大的磁各向异性。因此,这项研究为预测三角双锥构型Co(Ⅱ)配合物的磁各向异性提供了一个模型。Micro-SQUID测试表明两例配合物均表现出磁滞回线现象,证实它们都是单分子磁体。
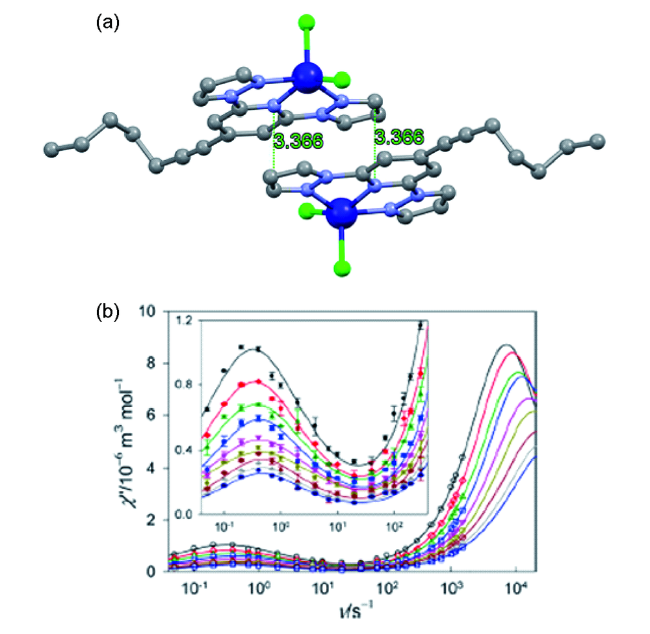
叠氮配体是研究磁化学最常用的配体之一。Schweinfurth等利用叠氮配体合成了一个新型五配位的单核Co(Ⅱ)配合物[Co(tbta)(N3)]ClO4·3CH3CN(tbta=tris-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-amine),并研究了磁性质[52]。该配合物具有单轴磁各向异性(D=-10.7 cm-1)和显著的横向零场分裂能(E/D=0.22),在3000 Oe直流场下表现出单离子磁体性质。
五配位的四方锥构型的Mn(Ⅲ)基单离子磁体只被报道过一次,而大部分的Mn(Ⅲ)基单离子磁体都是八面体构型。Dolai等基于salen配体合成了两例新型的四方锥构型的单核Mn(Ⅲ)配合物。两例配合物都具有负的零场分裂能,表现出场致慢磁弛豫现象[53]。
6 六配位的3d-SIMs
六配位铁基单离子磁体报道较少。Long课题组合成了六配位Fe(Ⅱ)的配合物[Fe(1-propyltetrazole)6](BF4)2[54]。该化合物在2000 Oe直流场的作用下产生了频率依赖的交流磁化率信号,表现出单离子磁体行为。而且在施加外场作用下,利用光激发可以使得该化合物的自旋态在S=0和S=±2之间发生转换。因此,当利用磁开关和磁光克尔效应作为读写机制时,这种三稳态就可以被用作分子三元信息存储。Sato等利用多酸配体得到一例严重扭曲八面体构型的Fe(Ⅲ)基单离子磁体,这是第一例高自旋Fe(Ⅲ)(S=5/2)的单离子磁体[55]。近期,Minato等也报道了多酸配体的基于高自旋Fe(Ⅲ)(S=5/2)基单离子磁体,并研究了多酸中杂原子对磁各向异性的影响[56]。
而在Co(Ⅱ)基单离子磁体中,六配位构型吸引了众多的关注。2012年,Vallejo等报道了首例六配位Co(Ⅱ)单离子磁体cis-[CoⅡ-(dmphen)2(NCS)2]·0.25EtOH(dmphen=2,9-dimethyl-1,10-phenanthroline)[57],当分别施加外场1.0、2.5和5.0 kG时,该化合物表现出单离子磁体行为,并得到相应的有效能垒为16.2~18.1 cm-1。EPR、磁性数据、理论计算表明该化合物的零场分裂能D> 0。该研究认为有效能垒值与横向零场分裂E相对应,因此横向零场分裂E代替了轴向零场分裂D导致了该化合物产生慢磁弛豫行为。
Colacio等也报道了一例具有D> 0的六配位Co(Ⅱ)基单离子磁体[Co(μ-L)(μ-OAc)Y(NO3)2](H2L=N, N',N″-trimethyl-N,N″-bis(2-hydroxy-3-methoxy-5-methylbenzyl)-diethylenetriamine)[58]。根据直流磁化数据、非弹性中子散射(INS)和X波段电子顺磁共振谱(X-EPR)测试表明该化合物的零场分裂能D为正值。由于存在量子隧穿效应(QTM),该化合物在外加场1000 Oe的情况下表现出缓慢的磁弛豫行为。该研究认为在这例单离子磁体中拉曼过程是主要的弛豫过程。Micro-SQUID测试进一步证实了该化合物是一例单分子磁体。
吴大雨等合成了两个类似的CoⅢ-CoⅡ-CoⅢ混合价态的配合物[(L1)4Co3(H2O)2](NO3)4·CH3OH·5H2O和[(L2)4Co3(H2O)2](NO3)4·6H2O[59]。据HF-EPR表征,这两种配合物的零场分裂能D都是正值。交流磁化率表明,第一个配合物并没有表现出单分子磁体行为,而第二个配合物在外加磁场1000 Oe下表现出缓慢的磁弛豫。化合物[Co(abpt)2(tcm)2](abpt=4-amino-3,5-bis(2-pyridyl)-1,2,4-triazole;tcm=tricyanomethanide anion)[60]和Co(oda)(aterpy)](oda2- =oxodiacetate;aterpy=4'-azido-2,2':6',2″-terpyridine)[61]被发现都具有正值的零场分裂能,并且都表现出单离子磁体行为。谢齐威等[62]利用双酚氧桥将Co(Ⅱ)与抗磁离子Y(Ⅲ)桥联,得到双核异金属配合物。磁性离子Co(Ⅱ)处于扭曲八面体配位环境中。该配合物表现出场致单离子磁体行为。宋友课题组发现了一例八面体构型的Co(Ⅱ)基单离子磁体具有双弛豫现象[63]。
本课题组合成了两例八面体构型的单核配合物[Co(L)4(NO3)2](L=3-methylpyridine和3-phenyl-pyrazole),其空间构型为扭曲的八面体,直流磁化率及HF-EPR测试表明了配合物具有较大的正值零场分裂能。在外加场的情况下两例配合物能表现出慢弛豫行为。其他文献报道的大多数八面体的Co(Ⅱ)基单离子磁体由多齿配体构筑,而这项研究中,所用的配位均为单齿配体,从而减少八面体的扭曲程度[64]。
有趣的是,八面体构型Co(Ⅱ)基单离子磁体中大部分配合物的零场分裂能D都是正值。Gómez-Coca等报道一例八面体构型Co(Ⅱ)配合物[Co(acac)2(H2O)2](acac=acetylacetonate)的磁性质,通过对其研究解释了这类具有正值零场分裂能(D> 0)配合物的单离子磁体行为[65]。他们认为Kramers离子的时间反演对称性抑制了直接自旋-声子过程导致慢磁弛豫的发生。另外,他们还提出核自旋与电子自旋的相互作用提供了新的磁弛豫通道。
然而,在八面体构型中Co(Ⅱ)基单离子磁体也出现了几例D< 0的化合物。本课题组利用咪唑合成了两个具有相同配阳离子的单核Co(Ⅱ)配合物[Co(imidazole)6][BPh4]2∙0.3CH3CN 和[Co(imidazole)6][NO3]2。在这两个配合物中由于抗衡阴离子的不同,从而导致八面体构型的配阳离子[Co(imidazole)6]2+产生理想的Ci和D3d的两种空间对称。
近年来,三棱柱构型的Co(Ⅱ)基单离子磁体被频繁研究。令人诧异的是,在这种配位环境下,单离子磁体均表现出单轴磁各向异性(D < 0),而且它们均具有较高的有效能垒。2013年,高松课题组报道了一例三棱柱构型的Co(Ⅱ)Co(Ⅲ)3配合物(HNEt3)+ (CoⅡCo3ⅢL6)-(H2L=R-4-bromo-2-((2-hydroxy-1-phenylethylimino)methyl)phenol)[68]。如图7所示,该化合物的Co(Ⅱ)离子与6个O原子配位,形成了稍微扭曲的三棱柱配位构型。磁性数据以及理论计算表明该化合物具有大的而且负的零场分裂能D。该化合物在不需要加场的情况下就能表现出慢磁弛豫现象,而且Ueff达到了109 K。Novikov等利用三吡咯肟盐合成了两例三棱柱构型的Co(Ⅱ)基单离子磁体,其具有非常大的轴各向异性,在加场的情况下其中一例配合物的有效能垒可达152 cm-1[69]。最近,又有具有三角反棱柱构型的Co(Ⅱ)基单离子磁体表现出单轴磁各向异性(D < 0)的报道[70,71]。这些研究结果表明在六配位构型的单核Co(Ⅱ)配合物中零场分裂能(D值)的符号和空间构型具有关联性。Gomez-Coca等利用理论计算预测了六配位的高自旋Co(Ⅱ)(S=3/2)配合物的零场分裂能符号,结果表明三棱柱构型则会产生负值的零场分裂能,八面体构型则两种可能性都有[72]。

图7 (HNEt3)+(CoⅡCo3ⅢL6)-分子结构图(a)、能级分裂图(b)、三棱柱构型示意图(c)和虚部交流磁化率(d) [68]Fig. 7 (a) The molecular structure of(HNEt3)+(CoⅡCo3ⅢL6)-;(b) The simplified d7 electron configuration;(c) The coordination model of the central Co(Ⅱ) ion;(d) variable-frequency out-of-phase ac susceptibility data; inset, Arrhenius analysis of the relaxation processes [68] |
在八面体构型的单核Ni(Ⅱ)配合物中,其零场分裂能D的符号与其构型的畸变密切相关,当八面体轴向压缩时D < 0,而轴向拉长则D > 0。Lomjanský等报道了一例具有轴向压缩的八面体结构的Ni(Ⅱ)基单离子磁体[Ni(NCS)2(nqu)2(H2O)2]2·nqu[73]。Miklovi$breve{c}$等利用2,6-二羧酸吡啶和2,6-二羟甲基吡啶配体合成了六配位的Ni(Ⅱ)基单离子磁体[Ni(pydc)(pydm)]·H2O。该配合物具有轴向压缩的八面体构型和零场分裂能D < 0,表现出双弛豫过程[74]。此后,他们将Ni(Ⅱ)换成Cu(Ⅱ),获得了一例扭曲八面体构型的单核Cu(Ⅱ)配合物。磁性表征说明该配合物的S=1/2,具有场致慢磁弛豫现象。这是首例Cu(Ⅱ)基离子磁体[75]。
基于Mn离子的单分子磁体一直以多核化合物为主,而对于单核Mn离子的单分子磁体鲜有报道。在八面体构型中,受杨-泰勒畸变的影响,高自旋Mn(Ⅲ)离子的基态谱项5Eg通常分裂成5A1g和5B1g,当八面体呈轴向伸长畸变时,Mn(Ⅲ)的基态为5A1g,导致零场分裂能D< 0;当八面体呈轴向压缩畸变时,Mn(Ⅲ)的基态为5B1g,导致零场分裂能D> 0[76]。因此,产生D< 0的伸长型八面体构型的单核Mn(Ⅲ)化合物具备成为单离子磁体的可能。目前报道的Mn(Ⅲ)基单离子磁体几乎都为八面体构型。Ishikawa等利用抗磁单元[CoⅢ(CN)6]构筑了Mn(Ⅲ)-salen型的配合物 [Mn(5-TMAM(R)-salmen) H2O)-Co(CN)6]·7H2O·MeCN(5-TMAM(R)-salmen=(R)-N,N'-(1-methylethylene)bis(5-trimethylammoniomethyl salicylidene-iminate))[77]。该化合物在加场的情况下只是表现出虚部交流磁化率的频率依赖行为,并没有出现相应的峰值。通过Kramers-Kronig模型ln(χM″/χM')=ln(ωτo) + Ueff/kBT可以拟合交流磁化率数据得到有效能垒为9.3 cm-1。与之类似的还有Sanakis等报道的Mn(Ⅲ)单离子磁体[Mn{(OPPh2)2N}3][78]和Sato等利用多酸配体得到的八面体构型Mn(Ⅲ)单离子磁体TBA7H10[Mn(A-α-SiW9O34)2]∙3H2O[55]。2013年,Vallejo等观测到一例八面体的Mn(Ⅲ)单离子磁体(Ph4P)[Mn(opbaCl2)(py)2](H4opbaCl2 =N,N'-3,4-dichloro-o-phenylenebis(oxamic acid), Ph4P+ =tetraphenylphosphonium cation)[79]。当施加外场1.0 KOe,该化合物在频率1~10 000 Hz内表现出慢磁弛豫行为(图8)。HF-EPR、磁化数据以及理论计算都得到该化合物的零场分裂能D< 0。通过Micro-SQUID测试,该化合物出现磁滞回线现象。2015年,Craig等也报道了一例伸长型杨-泰勒畸变的配合物Na5[Mn(L-tart)2]·12H2O(L-tart=L-tartrate)[80]。经HF-EPR表征,该化合物具有负值的零场分裂能D=-3.23 cm-1和较小的横向零场分裂E=0.032 cm-1。交流磁化率表明当施加外场时该化合物呈现出慢磁弛豫现象。

本课题组研究了三例基于二苯甲酰基甲烷(dbm-)的六配位的单核Mn(Ⅲ)化合物[Mn(dbm)3],[Mn(dbm)2(L)2](ClO4)(L=DMSO,py)的磁性质[81]。三个配合物结构都呈现出杨-泰勒畸变的伸长型扭曲,并具有负值的零场分裂能D,证明了伸长型杨-泰勒畸变的结构具有单轴磁各向异性。在施加直流场下三例配合物都表现出单离子磁体行为。其中一例配合物的有效能垒值Ueff达到了18.5 cm-1,明显高于其他的Mn(Ⅲ)基单离子磁体。
由于Mn(Ⅱ)离子的S=5/2和L=0,所以这种缺少自旋-轨道耦合作用的离子一般不存在磁各向异性,因此Mn(Ⅱ)不会被用来构筑单离子磁体。然而,Benniston等发现了一例Mn(Ⅱ)基单离子磁体[89]。它是由[Mn(bipy)2OH2]2+单元分布在羰基钙形成的长链上构成。二价Mn离子与两个联吡啶的4个N,二氯乙酸的羰基中的1个O和1个H2O进行配位形成了八面体配位构型。直流磁化率表明该配合物的零场分裂能非常小,约为10-2数量级。但是,在这个配合物中依然观察到了场致双弛豫现象。在低频区域的弛豫属于直接过程,而高频区域的弛豫过程归属于一种共振声子捕获机制。
7 高配位数(七配位和八配位)的3d-SIMs
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
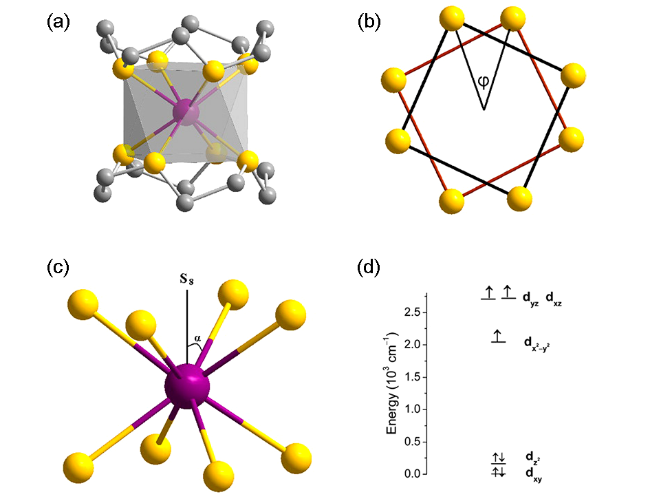
图9 [Co(12-crown-4)2]2+的结构示意图(a)、四方反棱柱上平面与下平面形成的扭转角φ示意图(b)、中心轴与配位键所形成的α示意图(c)、d轨道分裂图(d) [90]Fig. 9 (a) Side view of molecular structure of the cation [Co(12-crown-4)2]2+;(b) Twist angle φ defined as the rotation angle of one coordination square away from the eclipse conformation to the other;(c) The angle α defined as the eight metal-ligand bonds make with the S8 axis passing through the metal atom Co;(d) Electronic configuration and d-orbital energy level diagram for the molecule from DFT calculation [90] |
高松课题组合成了一例八配位的单核Fe(Ⅱ)配合物[Fe(L1)2](ClO4)2(L1=1,10-phenanthroline-2,9-dicarboxylic acid)[91]。在该配合物中,[FeN4O4]配位构型呈严重扭曲的十二面体。在1.4 kOe外加磁场下,此配合物具有慢磁弛豫行为。这是首例八配位的铁基单离子磁体。
本课题组合成了两例七配位的Co(Ⅱ)基单离子磁体[Co(L)3(NO3)2](L=4-tert-butylpyridine; iso-quinoline)[94]。结构表征显示其为五角双锥构型。HF-EPR证实这两例配合物具有易面磁各向异性(D> 0)。另外,Habib等也得到了具有易面磁各向异性的五角双锥构型的Co(Ⅱ)基单离子磁体[95]。为了研究其弛豫机理,加入抗磁离子Zn(Ⅱ)进行磁稀释。 研究发现,[Co(DAPBH)(NO3)(H2O)](NO3)配合物表现出两个弛豫过程,其中一个弛豫过程随着磁稀释样品中钴含量的减少而逐渐消失,而另外一个弛豫过程存在于不同稀释程度的磁稀释样品中。因此,其中一个弛豫过程是由于施加的直流场太大导致分子间发生相互作用而产生的;而另外一个弛豫过程是由含磁性离子Co(Ⅱ)单元固有的单分子磁体性质产生的。
这种五角双锥构型在二价铁的单离子磁体中也被发现。Sutter课题组利用超分子自组装合成了铁镍双金属的链状配合物,其中[Ni(Ⅱ)(CN)4]2-呈平面四方形,表现出抗磁性[96]。Fe…Fe之间的最短距离为8 Å。由于这种超分子组装改变了晶体堆积的密度,抑制了自旋晶格的弛豫过程,使其产生了热激发的Orbach过程,从而产生了单离子磁体性质。
8 理论研究
在分子磁性材料研究中,随着实验研究的快速发展,理论研究方面也取得了很大的进展。由于实验仪器等表征手段的限制,理论计算分析已成为3d过渡金属单离子磁体研究必不可少的手段。在前述的实验研究进展中,理论计算作为重要的辅助手段经常被用来分析金属离子的d轨道能级分裂以及电子布局和旋轨耦合,从而理解和探究磁各向异性(D和E)的起源。重要的是,在研究磁构关系时进行理论计算是非常有必要,用于定性分析配体原子、配位键长、键角及配体场的对称性等对磁各向异性的影响,从而对单离子磁体进行预测和筛选,指导高性能的单离子磁体的设计合成。
Ruiz课题组利用完全活性空间自洽场方法-受限制空间相互作用(CASSCF-RASSI)对[Fe(tpaR)]-体系进行了磁各向异性的分析[98]。该研究探讨了配体tpa的不对称性、Fe(Ⅱ)离子偏离三角单锥底面的距离和配体N的碱度与磁各向异性的关联性。计算表明Fe(Ⅱ)离子偏离三角单锥底面的距离对磁各向异性影响最大,偏离度越大则D值减小而E值增大。同时,小的配体碱度也能减弱零场分裂能。
Neese课题组开发了计算分子磁性的Orca软件,并基于分子磁性理论研究做出了许多的重要工作。直线型的单核配合物一般都具有良好的单离子磁体性能。2013年他们利用完全活性空间自洽场方法(CASSCF)、N-电子价微扰理论(NEVPT2)和准简并微扰理论(QDPT)对文献[13]中的一系列直线型Fe(Ⅱ)基单离子磁体的化学键、电子振动耦合和磁各向异性进行了理论研究[99]。结果表明,在所有的化合物中3 和4s轨道发生强烈的混合;除了配合物Fe(OAr’)2,在其他化合物中较强的π给电子配体产生了π键各向异性;化合物Fe(OAr’)2中Fe—O的σ-π轨道混合。虽然这些特异的化学键导致了基态5Δ(3dxy, 3 )发生对称性的降低和分裂,但是一阶自旋-轨道耦合对系统的影响更大,产生了重要的轨道贡献。而这种自旋-轨道耦合往往受配位几何扭曲的影响。
Co(Ⅱ)基单离子磁体是过渡金属单离子磁体中数量最多的一种。首次被发现的Co(Ⅱ)单离子磁体属于四面体构型。Neese课题组采用理论计算对这种体系(PPh4)2[Co(XPh)4](X=O, S, Se)进行了磁构关系的研究。理论计算采用了从头算的配体场理论[100]。在探究磁构关系时主要考虑了金属-配体的共价键、自旋-轨道耦合和配位几何的畸变。结果证实随着配位原子从O到Te的改变,四面体的配体场减弱,而零场分裂能D的绝对值增大。而且虚拟的配合物[Co(TePh)4]2-的D值大于配合物(PPh4)2[Co(SPh)4]的两倍。
在以上的理论计算文献中主要探讨磁各向异性与结构的关系,而Lunghi等系统研究了在单离子磁体[(tpaPh)Fe]-中自旋-声子耦合导致自旋弛豫的行为[103]。从头计算结果表明在稀释的晶体的自旋-声子慢弛豫过程中声频声子并不活跃。而且,分子内振动模式产生的各向异性张量调节的数量级要高于这些相关的旋转。旋转和内部振动是产生弛豫行为的主要原因。因此,除了自旋,晶格动力学也是设计合成单离子磁体需要考虑的因素。
9 结论与展望
在过去二十多年中,单分子磁体作为分子磁体领域最重要的发现之一,已吸引了众多研究者的广泛关注。具有较强磁各向异性的3d过渡金属单离子磁体是近年来兴起的研究领域之一。研究者们成功构筑出从二配位到八配位的3d过渡金属单离子磁体,其构型包括:直线型、平面三角形、四配位的四面体和三角单锥、五配位的四方单锥和三角双锥、六配位的八面体和三棱柱以及三角反棱柱、七配位的五角双锥、八配位的四方反棱柱和十二面体。这些研究表明:首先,二配位的直线构型有利于形成大的单轴磁各向异性,因此这类配合物具有相当高的弛豫能垒,超过了经典的Mn簇单分子磁体的能垒;其次,绝大部分3d过渡金属单离子磁体的配位数为二配位到六配位,仅有几例七配位和八配位的单离子磁体被报道,证实了低配位数的配体场确实有利于获得单离子磁体,而在高配位的构型中采用弱配位能力的配体也能够构筑出单离子磁体;最后,无论从实验层面还是理论层面,已初步获得了一定的磁构关系,如配位原子、配位构型对磁各向异性的影响。
然而,随着研究深入,3d过渡金属单离子磁体还面临一些挑战:首先,3d过渡金属单离子磁体的有效能垒较低,难以超越性能优异的稀土基单分子磁体;并且其阻塞温度远远低于液氮温度,很难应用于实际。增强磁各向异性和抑制量子隧穿效应是提高有效能垒和阻塞温度的有效方法。考虑配体构型的轴对称性,选择能够形成弱配体场的配体,构筑出具有大的单轴磁各向异性的单核配合物,一直是获得高性能单分子磁体的一种有效途径。其次,许多3d过渡金属单离子磁体的磁各向异性和慢弛豫机理仍然得不到合理的解释,如Co(Ⅱ)基单离子磁体的D> 0,许多解释还停留在理论计算层面,实验的表征仍然比较匮乏。最后,多功能性协同效应的3d过渡金属单离子磁体还鲜有报道,如一例六配位的[Fe(1-propyltetrazole)6](BF4)2在施加外场作用下可利用光激发使自旋态在S=0和S=±2之间发生可以转换[54],像这种具有光磁效应的单离子磁体以及其他多功能的分子磁体[104]都有可能成为该研究领域的重要课题。