



Contents
1 Introduction
2 Preparation of magnetic nanocatalysts
2.1 Magnetic core
2.2 Protective surface coating of magnetic core
2.3 Loading of nano-components and its catalytic application
3 Recent advances of magnetic nanocatalysts
3.1 Recent advances of magnetic nanocatalysts in the fields of catalytic desulfurization
3.2 Recent advances of magnetic nanocatalysts in the fields of catalytic conversion of biomass to chemicals
3.3 Recent advances of magnetic nanocatalysts in the fields of production of biodiesel
3.4 Recent advances of magnetic nanocatalysts in the fields of coal liquefaction
4 Conclusion and outlook
1 引言
催化剂是“绿色化学”的重要组成部分,现代化学家面临的挑战之一是如何更有效地制备及应用环保型催化剂。“绿色”催化剂必须具备特定的特征,包括制备成本低、活性高、选择性好、稳定性高、回收效率高、可回收性好等。研究表明,作为半异相催化剂,纳米催化剂具有更高的催化活性[3]。纳米颗粒凭借其独特性能如:比表面积大、暴露于表面的配位不饱和活性位点多、无毒、环保等特点在各个领域具有广泛应用[4]。但纳米催化剂也存在诸多缺陷,由于尺寸小,纳米尺寸的催化剂采用过滤、离心、膜分离等传统方法很难从反应体系中分离出来。通过将具有优异磁学性能的磁性纳米粒子与催化性能相结合制备的磁性纳米催化剂,在外加磁场作用下可以快速分离,同时具备高催化活性和易分离的特点,是未来催化剂发展的重要方向[5,6,7,8]。本文将对磁性纳米催化剂的合成方法及结构性质做相应的介绍,并对该研究方向的发展作了展望。
2 磁性纳米催化剂的制备
磁性纳米催化剂的结构由具有超顺磁性的无机磁核、保护磁核的表面修饰、提供负载活性纳米组分担载的壳层材料组成,本文分别介绍各组成的制备方法。
2.1 磁核
磁性纳米催化剂磁核的制备应该从价廉易得的材料开始,并从可重复性和可扩展性着手。理想的磁核还应具有高磁化率和低毒性特性,同时,可通过惰性但易改性的壳层来保护其在恶劣的反应条件下的稳定性并促进其进一步的官能化[9]。
磁性纳米颗粒的制备方法影响着颗粒形态(尺寸、形状、分布)、组成、磁性、表面化学和催化应用方面。常用的制备方法有共沉淀法、微乳液法、溶胶-凝胶法、喷雾和激光热解法、水热反应法、超声分解法、微波辐射法、生物合成法等[3, 10~15]。可选作磁核的材料有金属(Fe、Co、Ni等),合金(Fe-Co、Fe-Ni等),铁氧化物(FeO、γ-Fe2O3、Fe3O4等)或尖晶石铁氧体MFe2O4(M=Co、Mn、Cu、Zn、Fe、Ni、Mg等)。五羰基合铁[Fe(CO)5]是合成铁纳米颗粒最常用的前驱体[16]。由于金属铁在空气中极易氧化,合成纯金属纳米颗粒是一项艰巨的任务,因此至少在这种颗粒的表面包覆几种天然氧化物。在众多的铁氧化物中,因为FeO对空气更敏感,所以γ-Fe2O3和Fe3O4比FeO受到更多关注。
含MFe2O4(M=Co、Mn、Cu、Zn、Fe、Ni、Mg等)的尖晶石铁氧体由于其高催化活性、极低的溶解度、稳定的晶体结构,特别是其强磁响应性等而被广泛应用[17]。Chandel等[18]把各种尖晶石铁氧体纳米粒子MFe2O4(M=Co、Ni、Cu、Zn)应用于Biginelli催化反应中,使用基于水溶液的新方法合成所有铁氧体纳米颗粒,其催化活性遵循CoFe2O4>CuFe2O4>NiFe2O4>ZnFe2O4的降序排列。这些纳米颗粒可直接用作催化剂,不需要表面改性或官能化。通过外加磁体,这些铁氧体纳米颗粒在反应后易与反应混合物分离。与其合成方法不同的是,Quandt等[19]通过旋涂金属硝酸盐在N,N-二甲基甲酰胺和乙酸中的溶液,随后在不同温度下加热,在铂涂覆的硅晶片上制备MFe2O4(M=Co、Mg、Cu)薄膜。在制备过程中,这些薄膜具有各向异性微晶生长的特性。而Patnaik等[20]通过原位煅烧,然后通过物理吸附和原位氧化聚合方法加载PANI,制得聚合物敏化的g-C3N4/ZnFe2O4光催化剂。其易于分离,并且可以有效去除废水中的有毒污染物如Cr(Ⅵ)和苯酚。这种基于磁性的半导体光催化剂克服了分离和二次污染的问题。由于不使用额外的清除剂,因此该方法对于环境修复开辟了新的方向。
上述磁核中,Fe3O4合成方法简单,比饱和磁化强度较高,得到了人们的广泛关注。磁性载体受到人们关注的原因有:第一,磁性载体带有磁性,催化反应结束后,外加磁场存在的条件下,磁性催化剂就能简单地与反应体系分离,避免了过滤、离心等其他繁琐的步骤;第二,去掉外磁场的作用,通过简单的震荡或者搅拌,超顺磁性的磁性纳米粒子又可以均匀分散在反应体系中。因此反应中引入磁性载体可以轻松地实现催化剂与反应体系的快速分离,同时,可以提高催化剂的重复使用性。但是,裸露的磁性纳米粒子在催化反应时容易发生团聚现象,而且容易被溶液中的酸碱物质腐蚀,导致催化剂不能均匀地分散在催化体系内、催化活性降低、增加反应成本[21, 22]。即使在水介质中,由于吉布斯自由能和一些磁性纳米粒子的疏水性也会导致磁核的团聚[23]。为了解决这一问题,需要对磁核进行保护处理,一般采用合适的稳定配体或涂层材料修饰磁性纳米颗粒。
2.2 磁核的表面修饰
由于处理温度对磁核磁性有很大影响,且为避免磁核与负载的活性组分发生反应及磁核间的团聚现象,同时维持磁核在催化反应中的稳定性,常对磁核进行表面修饰,以满足不同的催化环境。传统的方法主要是利用稳定配体或涂层材料(包括小分子、二氧化硅、聚合物、碳、金属或金属氧化物纳米粒子以及它们的逐层组合)修饰磁性纳米颗粒。此外,也进一步直接将活性催化单元以化学键或者沉积作用与包覆材料相互作用负载在磁性载体上。
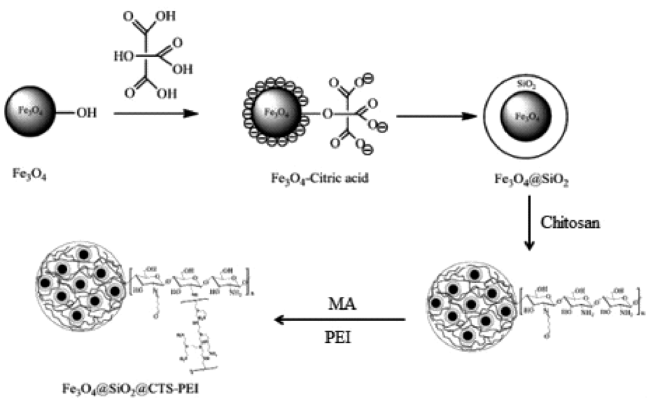
在各种包覆材料中,二氧化硅因其稳定性高、表面功能化简单等特性而使用最为广泛。包覆二氧化硅较常用的方法为溶胶-凝胶法和微乳液法,其中溶胶-凝胶法包覆程序简单且无需添加表面活性剂,然而采用这种方法合成的核-壳结构还有待确认[24]。溶胶凝胶法主要采取Stöber法实现,其通过硅醇盐前体在碱性醇-水混合物水解和缩合过程形成二氧化硅球。Wierucka等[25]证明了溶胶-凝胶Stöber方法可用于通过在水、氨和2-丙醇混合物中水解硅酸四乙酯(TEOS)来包覆具有均匀二氧化硅层的赤铁矿颗粒。溶胶-凝胶法的主要缺点是磁性纳米粒子在醇-水混合物中稳定性差,通常在二氧化硅沉积之前沉淀。De Mongolfier等[26]使用溶胶-凝胶法进行二氧化硅涂覆之前,通过用硅酸钠改变磁铁矿纳米粒子的表面来防止絮凝。通过将TEOS添加到含有表面改性的磁铁矿纳米颗粒的乙醇和氨的混合物中来进行二氧化硅包覆。微乳液法可较好地控制核-壳形态,其依赖于TEOS分散在表面活性剂中的预合成或共沉淀的磁性纳米粒子周围的水解和缩合。球形二氧化硅通过氨催化的TEOS水解形成。理论上,纳米颗粒的尺寸和二氧化硅壳的厚度可通过改变微乳液配方来调节,例如选择表面活性剂,然而,在实践中,很难控制颗粒的最终尺寸。孙西同等[27]首先利用化学共沉淀法制备柠檬酸修饰的磁性Fe3O4纳米颗粒,然后分别通过硅酸钠水解法和溶胶凝胶法制备单颗粒包覆磁性SiO2纳米颗粒,最后以壳聚糖作为壳层材料,利用溶胶凝胶法和乳液交联法制备了聚乙烯亚胺改性磁性壳聚糖微球(Fe3O4-SiO2-CTS-PEI)(图1)。实验表明,Fe3O4-SiO2-CTS-PEI催化剂表现出高耐酸性和磁响应性,且对Cr(Ⅵ)的吸附容量高达236.4 mg/g;经过五次吸附-解吸循环后,发现Fe3O4-SiO2-CTS-PEI可以有效再生并重新进行Cr(Ⅵ)的吸附,并可在外磁场作用下快速分离回收。
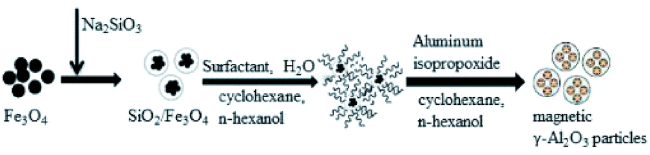
Al2O3作为包覆材料的磁性纳米颗粒的研究较少。郎宇琪等[28]采用微乳液法合成了平均粒径200 nm左右的γ-Al2O3/SiO2@Fe3O4磁性复合纳米催化剂(图2),然后在该催化剂上负载Pd,合成了Pd磁性纳米催化剂。该催化剂具有非常高的催化活性,在特定条件下硝基苯的转化率和苯胺选择性均可达到100%,并且该催化剂重复使用10次后仍可保持很高的催化活性,在外磁场作用下可快速分离回收。而Zong等[29]通过将NiSO4溶液浸渍到含有Fe3O4磁性材料的γ-Al2O3载体上制得磁性NiSO4/γ-Al2O3催化剂。在固定床和磁稳定床(MSB)反应器中评价磁性催化剂的轻质催化裂化汽油烯烃低聚反应。实验证实了MSB在制造具有高生产率和柔韧性的清洁柴油燃料的潜在价值。
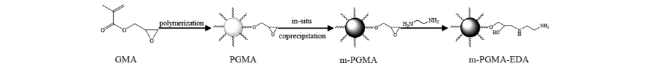
在磁性纳米颗粒表面引入具有官能团的聚合物(或枝状聚合物)是人们越来越关注的课题[8]。聚合物包覆材料可分为合成材料和天然材料。基于聚(乙烯-乙酸乙烯酯)、聚(乙烯基吡咯烷酮)(PVP)、聚(乳酸-乙醇酸)(PLGA)、聚(乙二醇)(PEG)和聚(乙烯醇)(PVA)的聚合物是合成聚合物系统的典型实例。天然聚合物体系包括明胶、葡聚糖、壳聚糖、普鲁兰多糖、淀粉等[30]。一般来说,用表面活性剂或聚合物包覆磁性纳米颗粒的方法不同于用无机氧化物包覆的方法。在这种情况下,聚合物或表面活性剂可以通过化学锚定或物理吸附在磁性纳米颗粒上形成单层或双层[31]。这种吸附产生排斥力(主要是空间排斥力),稳定磁性颗粒,避免沉淀。含有官能团的聚合物,如羧酸、磷酸盐和硫酸盐,适用于涂覆氧化铁基磁性材料。最常用的聚合物是聚(吡咯)、聚(苯胺)、聚(烷基氰基丙烯酸酯)、聚(亚甲基丙二酸酯)和聚酯,例如聚(乳酸)、聚(乙醇酸)、聚(ε-己内酯)及其共聚物。董婷婷等[32]成功合成了一系列具有不同胺密度的磁性聚(甲基丙烯酸缩水甘油酯)(m-PGMA-EDA)微球(图3)。并测定了他们对Cd(Ⅱ)的饱和吸附容量。实验表明,该磁性微球对Cd(Ⅱ)有一定的吸附能力。聚(甲基丙烯酸缩水甘油酯)(PGMA)由于其高空隙率、高化学稳定性、机械强度、活性环氧基团特性使其在涂料、印刷油墨、黏合剂以及半导体等领域均有应用。Altuntas等[33]报道了使用PGMA从人血液中纯化血红蛋白。他们利用高活性表面环氧基团将生物分子固定在PGMA表面。具有磁核和聚合物壳的磁性微粒的制备已成为生物学应用的重要课题[34]。与连续相载体相比,磁性微粒具有加工时间更短、化学和机械耐久性更高、成本更低等优点[35]。
多巴胺衍生物[36]、三乙氧基硅烷[37]、膦酸官能化分子[38]和谷胱甘肽[39]常用于实现磁性纳米催化剂的稳定化和功能化。该过程产生嫁接位点或反应位点以结合催化物质。多巴胺对磁性纳米颗粒中的铁离子具有出色配位能力,通常通过超声处理悬浮液中的混合物来促进配位。Sato等[40]报道了第一个相转移催化剂(季铵盐和鏻盐)修饰的磁性纳米颗粒的例子。在合成的过程中,(3-碘丙基)三甲氧基硅烷依次与季铵盐和鏻盐反应,然后使用磁性纳米颗粒锚定获得均相三乙氧基硅烷基官能化的相转移催化剂。这种半异相催化剂在苯酚钠(PhONa)与叔丁基溴(n-BuBr)在甲苯和水的混合溶剂中的O-烷基化反应显示出高活性和稳定性。最近,Peng等[41]开发出一种简单的两步法制备Fe3O4@PS/PDA-Ag纳米管,其尺寸可控且高度分散。首先使用含有高度分散的油酸改性的Fe3O4颗粒在聚苯乙烯(PS)/甲苯溶液中以商业AAO模板制备具有良好机械性能的Fe3O4@PS纳米管。接着,将多巴胺包覆在磁性(Fe3O4@PS)纳米管表面原位形成银纳米颗粒。通过一系列表征实验,发现制备的Fe3O4@PS/PDA-Ag纳米管具有较大的比表面积,在催化水相中NaBH4还原4-硝基苯酚(4-NP)具有极好的活性,且容易与流体分离并通过外部磁体回收。
碳材料具有更高的化学和热稳定性,并且对大多数化学品是不可渗透的,可完全屏蔽内部磁核。石墨碳层可提供有效的抗氧化和酸腐蚀屏障。此外,碳涂层更常用于金属状态的纳米颗粒,因此纳米材料具有比相应氧化物更高的磁矩。各种碳包覆的铁基二元合金(FeCu、FeCo和FeNi)通过连续喷涂、化学沉淀和高温下的可控热解制备[42]。然而,在对应反应条件下,Fe3O4易被还原为Fe0。碳化物碳涂层磁铁矿更难以获得。Tristão等[43]将Fe2O3的还原与使用甲烷的化学气相沉积工艺相结合,制备了磁性碳包覆Fe3O4颗粒。在600~900 ℃的温度下,用甲烷直接还原Fe2O3,主要生成包覆有高达4%非晶碳的Fe3O4颗粒。制备的催化剂在加氢脱氯反应中具有活性。Johnson等[44]采用另一种方法通过在氩气气氛下高温热解硬脂酸铁产生碳包覆的磁性Fe和Fe3C纳米粒子。得到的碳包覆磁性纳米颗粒在空气中400 ℃下仍能稳定存在。这种直接一步盐转化工艺有很多优点,可以扩大规模,然而,该方法制备的纳米颗粒尺寸分布较宽,直径范围从20 nm~200 nm。
纯铁、钴和镍纳米粒子在空气中化学性质不稳定并且容易氧化,限制了其的应用。用贵金属层涂覆这种磁性颗粒可保护颗粒免于氧化。贵金属在磁性纳米粒子上的沉积可以通过不同的反应例如微乳液法、沉淀法或氧化还原转移法发生[45]。Park等[46]报道通过回流Co纳米粒子胶体(约6 nm)和[Pt(hfac)2](hfac=六氟乙酰丙酮酸盐)的含有十二烷基异氰化物作为稳定剂的壬烷溶液中所获得的颗粒在有机溶剂中是稳定的且可再分散的。将反应副产物分离,分析为[Co(hfac)2],表明Co0和Pt2+之间的氧化还原转移反应驱使核-壳结构的形成。铂在钴核周围形成壳并且壳表面是通过十二烷基异氰化物封端分子稳定。Co-Pt核-壳纳米粒子的磁性研究表明其保留纯Co核的大部分磁性。该催化剂较经济地利用了铂原子的优点,因为只有外部原子可用于试剂,并且磁性钴核在催化剂的分离和再循环中起关键作用。
此外,一些金属氢氧化物和氧化物层用于磁性纳米颗粒的包覆也可提高磁性纳米颗粒的稳定性。例如二氧化钛和二氧化铈已广泛用作催化剂载体,并且在与磁性纳米粒子结合时可以找到广泛的应用。制备方法包括磁性纳米材料的直接包覆和硅酸化磁性纳米材料的后包覆。Álvarez等[47]报道了用二氧化钛涂覆磁铁矿的溶胶-凝胶法,当涂覆二氧化钛之前加入二氧化硅时获得最佳结果。Ye等[48]通过调节二氧化钛前体四丁基钛酸盐(TnBT)的用量,在二氧化硅包覆的磁铁矿上控制沉积二氧化钛制备了类似于豌豆的纳米结构。He等[49]报道了用尿素和Ti(SO4)2均匀沉淀法用二氧化钛直接包覆磁铁矿。使用尿素可控制TiO2前体的水解和缩合速率,尿素缓慢释放氨并控制OH-的生成,并因此均匀地覆盖磁铁矿纳米粒子。此外,也有研究通过种子辅助水热法制备了高度有序的TiO2包覆的Fe2O3核-壳磁性纳米颗粒。
2.3 活性纳米组分的担载及其催化应用
基于金属材料的催化研究是石油化工中的一个热点研究领域,因为金属材料容易激发反应底物的活性位点[50, 51]。在过去的几十年中,广泛研究了固定在载体(如二氧化硅、氧化铝、沸石和介孔材料)上的金属材料在各种化学反应中的催化活性[52,53,54]。虽然这些制备方法很有价值,但仍存在一些缺点,例如繁琐的后处理步骤、收益率低以及难以分离或重复利用。此外,载体有时催化副反应[52],催化剂表面上存在较少的活性位点可用于催化,反应性和选择性较低。因此,新载体材料的发展是现代催化科学的一个重要挑战。为克服这些缺点,最近,固定在磁性纳米颗粒上的金属材料作为有效的磁性可回收催化剂。目前,应用于催化领域的金属材料主要有Cu、Ni、Au、Pd、Rh、Ir、Mo及其他活性组分。
2.3.1 贵金属负载磁性纳米催化剂
目前,应用于催化领域的贵金属材料主要有Pd、Pt、Ru、Au、Rh、Ir等。这些贵金属负载的磁性纳米催化剂不但能保持贵金属的催化性质,而且赋予其磁分离特性[55]。
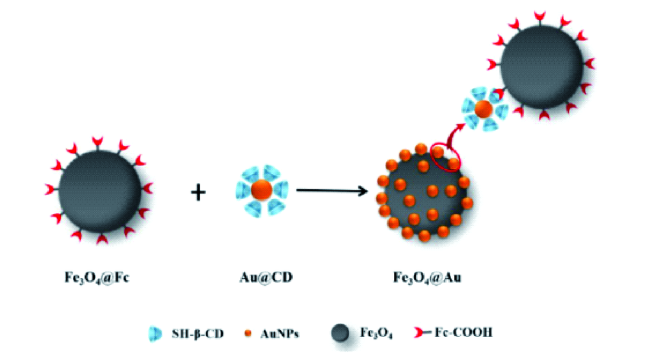
金包覆的磁性纳米颗粒成为近年来研究的焦点,用于磁控生物医学、分析和催化应用领域。然而,由于两个表面的不同性质,研究人员发现将贵金属直接包覆在磁性纳米颗粒表面是非常困难的。Ban等[56]合成了磁核约11 nm和约2.5 nm金包覆层的纳米颗粒。金作为一种选择性催化剂,可用于将醇氧化成相应的酮和醛也可以用于硝基苯的加氢反应中。在贵金属磁性催化方面,为了实现金纳米颗粒的回收及循环使用,屈虹男等[57]利用二茂铁修饰的四氧化三铁磁性纳米颗粒与β-环糊精修饰的金纳米颗粒自组装构建了Fe3O4@Au磁性纳米金催化剂(图4)。催化过程在25 ℃条件下进行,25 μg的Fe3O4@Au磁性纳米金催化剂可以在硼氢化钠存在条件下7 min内完成4-硝基苯酚的高效催化还原,转化率为99%,其催化速率高于具有类似结构的磁性纳米金催化剂。同时自组装磁性纳米催化剂具有良好的保存稳定性及循环使用性能,并可实现氧化还原响应性的可逆解组装过程,在其他贵金属磁性纳米催化剂的构建、生化分离、SERS检测等方面具有潜在应用价值。
钯因其在氧化反应中的高活性而著称,Pd纳米颗粒也常用于形成C—C键。Pd催化的Heck,Suzuki和Sonogashira偶联反应在理论和实践上均得到了广泛的研究[58],此外,在硝基苯加氢制苯胺的反应中钯催化剂也占有一席之地。郎宇琪等[28]系统研究了以W/O微乳液为纳米反应器的Pd金属组分负载的磁性纳米催化剂在硝基苯加氢制苯胺催化体系中的应用。通过改变水/表面活性剂比值、Pd负载量,成功制备了平均粒径7~20 nm均匀分布的Pd/磁性γ-Al2O3负载催化剂。在最佳实验条件下,硝基苯的转化率和苯胺选择性均可达到100%。实验表明该催化剂具有较好的稳定性和寿命,且具有超顺磁性,反应结束后可在外加磁场作用下与产物快速分离,实现催化剂回收和重复使用。
与Pd负载的磁性纳米颗粒类似,Pt负载的磁性纳米颗粒在还原和氢化反应中也有应用。Yamashita等[59]在油酸和油胺存在下还原乙酰丙酮铂(Pt(acac)2)制备Pd负载的磁性纳米颗粒。Pt原子位于壳层,而Fe原子位于核层。平均直径为2.5 nm的Fe-Pt纳米颗粒成功用于水系中4-NP的加氢反应。与在有机溶剂中相比,Fe-Pt纳米颗粒在水系中具有更好的催化活性,通过使用外加磁铁,纳米颗粒可以很容易地从水中分离回收。
2.3.2 过渡金属负载磁性纳米催化剂
目前,应用于催化领域的过渡金属材料主要有Ni、Mn、Ag等。镍纳米颗粒是一种很重要的磁性材料,除了其固有的磁性之外,还可以固定在基于铁氧化合物的磁性载体的表面上,以改善催化反应中的磁分离效率。负载镍的磁性纳米颗粒在纯水相中的使用研究较少,主要集中在硝基苯的加氢反应中。Shen等[60]利用原位还原法制备了一系列具有不同形貌的Ni官能化的磁性纳米颗粒(基于还原氧化石墨烯(GO/Ni)的磁性纳米镍复合材料)。该催化剂在NaBH4存在下在水中催化还原4-硝基苯酚显著增强,且很容易从反应系统中分离出来。另一种镍功能化的磁性纳米颗粒(钴镍合金纳米粒子)由Mandal等[61]报道,他们在非极性溶剂中通过湿法化学还原方法制备链状钴镍(CoNi)合金纳米粒子。纳米合金的成分可以通过改变前体Co和Ni的初始摩尔比实现。与Co和Ni纳米颗粒相似,Ni官能化的磁性纳米颗粒在Na2S2O3存在下K3[Fe(CN)6]的氧化还原反应中表现出良好的活性,NaBH4存在下在4-硝基苯酚还原中表现出良好的活性。此外,催化剂通过使用棒状磁体易于从反应混合物中回收,并且可以在没有进一步纯化的情况下有效地重复使用至少八次。
锰是常用的氧化反应催化剂。在卟啉配合物的研究方面应用较多,卟啉配合物是常用的生物类催化剂。Rezaefard等[62]报告了SiO2包覆的锰官能化的磁性纳米催化剂,并将其应用于水中烃类和硫化物的选择性氧化。锰官能化的磁性纳米催化剂通过使用胺作为连接物在二氧化硅包覆磁性纳米颗粒上的锰-卟啉络合物的协调锚定获得。该催化剂在水溶液中与四丁基过氧化硫代硫酸铵氧化烯烃和饱和烃具有很高的催化活性。类似地,硫化物也可以在水相中选择性地转化为砜,并在水-乙醇相中转化为亚砜。实验结果表明,在各种底物的氧化过程中,锰官能化的磁性纳米催化剂可以有效地回收和重复使用至少七次,产率几乎没有下降。
银作为一种传统的催化剂,使用非常广泛,也受到了很多关注。在NaBH4存在下银可以催化还原4-硝基苯酚(4-NP)。其催化作用的模型可以假定如下:首先,硼氢化物离子可逆地将表面氢物种转移到Ag表面,同时吸附4-NP;速率决定步骤是由表面氢还原4-NP;最终得到还原产物,完成催化循环。γ-Fe2O3纳米颗粒、二氧化硅包覆的γ-Fe2O3纳米颗粒和碳包覆的γ-Fe2O3纳米颗粒作为负载银的磁性纳米颗粒的载体均有报道。Shin等[63]最先报道了Ag负载磁性纳米颗粒的制备方法。他们通过在乙醇AgNO3溶液和丁胺体系中原位还原制备Ag负载的γ-Fe2O3纳米颗粒,将Ag纳米颗粒直接沉积在γ-Fe2O3纳米颗粒表面。在NaBH4存在下催化还原4-NP,在水中具有良好的催化活性。通过使用小型磁体可以容易地从溶液相中回收纳米颗粒。Jiang等[64]采用超声化学方法将Ag离子原位还原成Ag,以取代化学还原剂。该方法为制备Ag负载的磁性纳米颗粒提供了一种绿色高效的工艺。
2.3.3 金属氧化物或硫化物等负载磁性纳米催化剂
应用于催化领域的金属氧化物或硫化物等材料主要有铜配合物、铁氧化物、钴氧化物、锑氯化物、钨化合物、钼硫化物等。
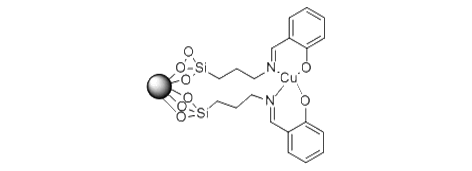
铁催化剂是氧化反应最常用催化剂之一。自含Fe酶的发现以来,针对铁催化剂的许多研究主要集中在仿生催化中,如含铁卟啉和席夫碱。然而,这些催化剂具有难分离、高成本、商品化困难等问题,其固定化引起人们的关注。因此,需要更简单、有效、安全和廉价的含铁催化剂。Shi等[66]报道了使用过氧化氢作为氧化剂的游离纳米Fe2O3在氧化醇和烯烃中的催化活性和选择性。粒径为20~50 nm的γ-Fe2O3对苯甲醛的转化率为33%,选择率为97%。当使用尺寸为3~5 nm的γ-Fe2O3时,可以达到更高的活性。此外,γ-Fe2O3纳米粒子的固有磁性使得催化剂可以简单地通过使用永磁体而容易与反应介质分离。
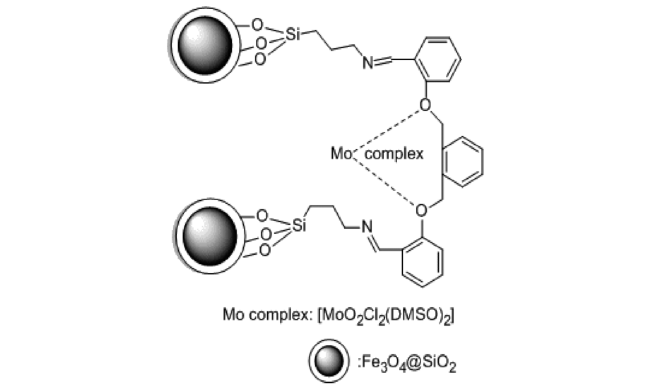
杂多酸在石油化工领域已被商业利用。虽然多数用于催化领域的杂多酸可以回收和再利用,但固定在磁性材料上的杂多酸只涉及12-钨磷酸(H3PW12O40)。最近,Rafiee等[67]报道了将H3PW12O40固定在二氧化硅包覆的γ-Fe2O3纳米颗粒表面上的杂多酸基负载W的磁性纳米颗粒。这些磁性纳米颗粒大多为球形,平均尺寸约为94 nm。磁性纳米颗粒表面活性位点比均相类似物更多。该特征在水相中的醛,胺和酮的一锅三组分Mannich型反应中具有高催化效率,产率高达98%。此外,催化剂可以在至少五个循环后回收和再利用,而其催化活性没有任何显著损失。
3 磁性纳米催化剂的应用进展
目前,磁性纳米催化剂在石油化工领域的应用主要包括在催化脱硫、生物质催化转化为化学品、生物柴油的制备、煤液化等。
3.1 磁性纳米催化剂在催化脱硫领域的应用
运输燃料中含硫化合物是最不受欢迎的污染物之一,它们可以通过燃烧转化成有毒的硫氧化物(SOx),对环境造成很大的污染。深度脱硫方法包括加氢脱硫(HDS)、氧化脱硫(DDS)、吸附脱硫、萃取脱硫等。将脱硫催化剂与磁性载体相结合,不仅可以延长催化剂使用寿命,降低生产成本,还有利于磁性纳米催化剂的回收和分离,因而受到越来越多的关注。
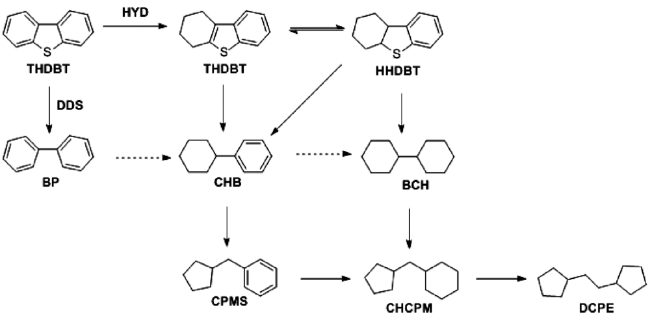
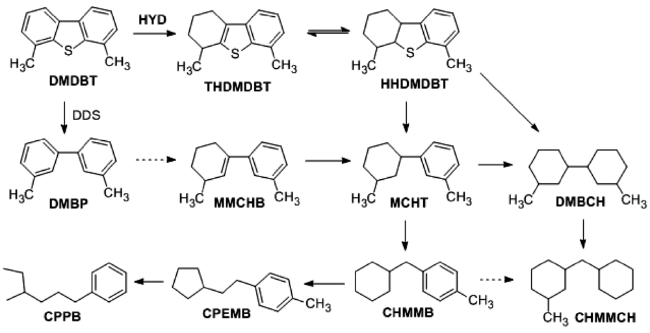
Zheng等[69]使用两种不同的表面活性剂(十六烷基三甲基氯化铵CTAC和十六烷基硫酸钠SDS)来辅助样品合成,制备了具有磁性和催化活性的双功能催化剂MoS2/SiO2/Fe3O4,该催化剂具有核-壳结构,磁核平均直径约为50 nm,在300 K的温度下MoS2/SiO2/Fe3O4颗粒的饱和磁化强度为59 emu/g。与不加表面活性剂的样品实验对比,发现表面活性剂对最终催化剂的性质和活性有很大影响。表面活性剂可以通过为晶体生长提供合适的位点来加速其前体生成MoS2的速度。催化剂的加氢脱硫(HDS)实验显示阳离子表面活性剂辅助制备的催化剂具有较高的活性。原因是SDS在合成温度200 ℃开始分解,在合成1 h内,几乎所有的SDS均分解,而TGA分析显示CTAC这种催化剂具有较强的磁性和较高的催化活性,并且易于通过磁场回收,使用寿命长,250 ℃才开始分解。Cat-CTAC在二苯并噻吩加氢脱硫中表现出最高的活性。然而,有人担心在含硫环境中使用这些复合材料以及作为含硫化催化剂的复合材料的稳定性。他们[70]又以Fe3S4为磁核,CTAC作为表面活性剂在催化剂层上作为活性相,外层由(Ni/Co)MoS2催化剂组成,然后以苯丙噻吩(DBT)和4,6-二甲基二苯并噻吩(4,6-DMDBT)作为模型含硫化合物测试加氢脱硫效果,并研究Ni和Co的促进作用。与MoS2/Fe3O4对比,NiMoS/Fe3S4对DBT和4,6-DMDBT具有较高的催化活性。DBT的HDS中CoMoS/Fe3S4的活性和该催化剂的DDS/HYD比率与Ni促进的催化剂类似。MoS2/Fe3S4显示出更高的HYD途径趋势,DDS/HYD比率为0.31。与MoS2/Fe3O4复合材料相比,MoS2/Fe3S4复合材料在HDS测试的几个循环中均表现出较高的稳定性,并且在前4个循环中表现出更高的加氢选择性。图7和图8分别为DBT和4,6-DMDBT的HDS可能路线。
与Zheng等的报道相比,Alizadeh等[71]发现,离子液体修饰的磁性纳米颗粒对油品也有一定的脱硫能力。与Zheng等制备方法不同的是,他们采用化学共沉淀法合成了平均粒径在10~15 nm的Fe3O4单分散纳米颗粒。然后采用Stöber法包覆SiO2,最后在该复合颗粒上负载了离子液体。在正己烷/乙腈双相体系中,使用H2O2作为氧化剂,测试了二苯并噻吩(DBT)作为模拟油的氧化脱硫效果。在最佳实验条件硫含量可分别从100 mg/L降低到2 mg/L。这些纳米磁性非均相催化剂可以通过施加外部磁场而容易地从反应混合物中分离出来并循环使用数次。
Wang等[72]利用两步法成功合成了具有核-壳结构的Cs2.5H0.5PW12O40/nano-Fe3O4/SiO2和H3PW12O40/ nano-Fe3O4/SiO2两种磁性可回收催化剂,用于催化燃料油的氧化脱硫。他们首先对磁核Fe3O4进行SiO2包覆,然后,将合成的具有Keggin结构的Cs2.5H0.5PW12O40或H3PW12O40担载到nano-Fe3O4/SiO2上,nano-Fe3O4/SiO2的平均直径约为300 nm。最佳合成工艺条件下,对燃料油CsPW/Fe3O4/SiO2 和HPW/Fe3O4/SiO2的脱硫率分别达到了91.4%和99.4%。当使用HPW/Fe3O4/SiO2时经氧化脱硫和连续萃取处理后的稳定汽油和催化柴油的脱硫率分别达到96.3%和97.1%。借助外部磁场可容易地回收催化剂,该磁性固体催化剂可多次重复使用,活性降低不大,且回收率较高。
氧化脱硫是深度脱硫、加氢脱硫的替代或互补技术[73],具有反应条件温和(常压低温)、选择性高、对二苯并噻吩(DBT)等空间位阻硫化合物有脱硫潜力等优点。在ODS过程中,芳香族硫化合物,如苯并噻吩(BT)、DBT及其烷基化衍生物被氧化成相应的砜或亚砜,可以使用有机萃取剂[74]除去。Rafiee等[75]将PW锚定在二氧化硅包覆的CoFe2O4磁性纳米粒子表面,制备了PW功能化磁性纳米粒子CoFe@Si-PW。该纳米颗粒成功用于含硫化合物的氧化。在温和的反应条件下,各种硫化物被选择性氧化成相应的砜。以H2O2为氧化剂,CoFe@Si-PW催化剂对DBT、BT、噻吩等模型油的ODS具有较高的催化活性。
3.2 磁性催化剂在生物质催化转化为化学品领域上的应用
生物质能已成为化石燃料的潜在替代品,生物质的催化转化是制备各种商品化学品或液体燃料的主要途径之一。然而生物质催化转化中常用的均相催化剂及非均相催化剂具有难回收再利用以及分离损失大等问题,限制了它们的应用。磁性纳米催化剂作为一种新型的催化剂,不仅可具有高催化活性,在外加磁场作用下还能实现催化剂的回收与重复利用,在工业生产得以连续化的同时,也降低了生产的成本,提高了生产效率。
碳水化合物是生物质的主要成分,被认为是生产生物燃料和平台化学品的理想原料。就生物质转化领域而言,磁性酸催化剂可以催化纤维素水解成葡萄糖,将果糖脱水成5-羟甲基糠醛(HMF),并将HMF醚化成5-乙氧基甲基糠醛(EMF)[76]。磁性金属催化剂已用于化学氧化和还原生物质基化学中间体[77]。最有潜力的应用之一是将基于C6的碳水化合物转化为HMF,HMF被认为是合成各种新产品的重要平台分子,以及替代化石资源衍生燃料和化学制品的重要中间体[78]。由果糖脱水合成HMF主要通过磁性磺酸催化剂进行催化。Wang等[79]制备了一种具有Fe3O4核和磺酸功能化二氧化硅壳的磁性酸催化剂(Fe3O4@Si/Ph-SO3H)。所制备的Fe3O4@Si/Ph-SO3H催化剂能有效催化果糖在二甲基亚砜(DMSO)中脱水成HMF,在100 ℃下反应3 h后果糖转化率为99%,HMF产率为82%。然而,在相同反应条件下,具有相同的H +量的常规Amberlyst-15催化剂仅产生16.7%的HMF,果糖转化率为60.1%。这些结果表明,Fe3O4@Si/Ph-SO3H催化剂显示出比Amberlyst-15催化剂更高的催化活性。除了优异的催化活性之外,该催化剂还可以磁性分离并循环数次而不会失去其活性。Martinez等[80]也报道了类似的工作,他们还使用磺化二氧化硅包覆的Fe3O4作为磁性酸催化剂,将木糖脱水成糠醛。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
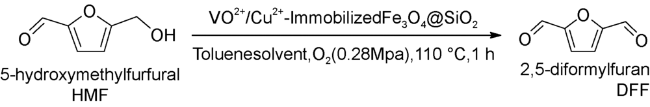
Karimi等[82]报道了一种通过使用磁性有机催化剂(Fe3O4@SiO2-TEMPO,MNST)将HMF氧化成DFF的新方法,其中2,2,6,6-四甲基哌啶氧化物(TEMPO)负载到Fe3O4@SiO2上。该催化体系需要使用亚硝酸叔丁酯(t-Bu-ONO)作为助催化剂并且使用乙酸作为添加剂。在1.013 KPa的O2,50 ℃下反应10 h,得到极好化学选择性(> 99%)的DFF。
3.3 磁性催化剂在生物柴油制备领域的应用
生物柴油是一种发动机燃料,可生物降解,无毒且环保。它不仅可以直接用作替代燃料,而且可以用作柴油燃料的清洁燃料添加剂。生物柴油通常在酸、碱或酶催化剂存在下通过植物油、大豆油或动物脂肪与醇的酯交换来制备。这些催化剂虽价格低廉但是对设备腐蚀性大,后处理复杂,同时也存在着分离、回收困难等的问题,进而制约了催化剂的进一步工业化应用。将具有优异磁性的磁性粒子引入到催化剂中,制备出磁性纳米催化剂,既可实现在外加磁场下对催化剂进行快速分离,又可保证纳米催化剂原有的催化效果。
可用于柴油生产的原料油种类较多,主要为大豆油(soybean oil)、乌柏油(stillingia oil)、棉籽油(cottonseed oil)、麻风树油(jatropha oils)等[83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98]。最近,许多研究者报道了通过磁性纳米催化剂生产生物柴油的研究。在磁性碱催化剂中,活性化合物CaO的使用在生产生物柴油方面受到越来越多人的关注。Fan等[83]以MFe2O4为磁核,采用水热法制备了一系列磁性CaO/MFe2O4(M2+)催化剂,应用于大豆油生产生物柴油的酯交换反应(表1,entry 1)。CaO/MFe2O4催化剂表现出较强的磁性和碱性。与CaO/ZnFe2O4和CaO/MnFe2O4相比,CaO/MFe2O4表现出较好的催化性能,较弱的吸湿性和较强的润湿性。在70 ℃下反应5 h后CaO/MFe2O4催化剂的生物柴油产率为87.4%。Liu等[84]用Na2CO3和NaOH作为沉淀剂将CaO负载在Fe3O4上制备了一种简单的磁性固体碱催化剂(CaO/Fe3O4),并研究了它对酯交换反应的催化活性(表1,entry 2)。实验发现,Ca2+与Fe3O4的比例影响催化活性,当Ca2+与Fe3O4的比例为7∶1时,催化活性最高,在甲醇/油摩尔比为15, 温度为70 ℃的条件下,80 min内生物柴油产率为95%,催化剂用量为2 wt%。他们还发现,纳米磁性固体碱催化剂的催化活性和回收率远远优于CaO。
| Entry | Substrate | Catalyst | Temperature(℃) | Time | Yield(%) | ref |
|---|---|---|---|---|---|---|
| 1 | soybean oil | CaO/CoFe2O4 | 70 | 5 h | 87.4 | 83 |
| 2 | soybean oil | CaO/CoFe2O4 | 70 | 5 h | 63 | 84 |
| 3 | soybean oil | K/BC-Fe2O3 | 60 | 60 min | 98 | 85 |
| 4 | jatropha oil | Zn8@Fe-C400 | 160 | 4 h | 100 | 86 |
| 5 | soybean oil | CaO/Fe3O4 | 70 | 80 min | 95 | 87 |
| 6 | stillingia oil | KF/CaO-Fe3O4 | 70 | 3 h | 95 | 88 |
| 7 | rapeseed oil | CaO/α-Fe | 60 | 2 h | 95.7 | 89 |
| 8 | soybean oil | Fe3O4/MCM-41 | 60 | 8 h | 99.2 | 90 |
| 9 | soybean oil | Fe3O4@HKUST-1 | 60 | 3 h | 92.3 | 91 |
| 10 | cottonseed oil | Na2O-SiO2/Fe3O4 | 65 | 100 min | 97 | 92 |
| 11 | soybean oil | LiFe5O8-LiFeO2 | 65 | 6 h | 96.5 | 93 |
| 12 | soybean oil | Fe3O4-MGO | 40 | 6 h | 92.8 | 94 |
| 13 | soybean oil | HAP-γ-Fe2O3 | 60 | 3 h | 99.6 | 95 |
| 14 | F platanifolia L.f. | Fe3O4@SiO2@SBA-15 | 85 | 5 h | 92.8 | 97 |
| 15 | K. integrifoliola | Fe3O4@SiO2(FS-BL-IL) | 160 | 10 h | 93.7 | 98 |
3.4 磁性纳米催化剂在煤液化方向的应用
直接煤液化(DCL)的煤液体作为“第二代生物燃料”越来越受到人们的重视。磁性纳米催化剂由于合成方法简单,合成材料廉价易得,并且易于分离,近年来备受关注。因此,如何在保持DCL高活性的同时使其易于分离成为科研人员关注的焦点。可以使用磁性分离技术来促进磁性DCL催化剂的快速回收,从而保持高DCL活性并且可以多次重复使用。
目前,将磁性纳米材料用于煤液化方面的研究正处于起步阶段,相关的研究较少。Traa等[99]采用少量的磁性纳米Co/SiO2/Fe3O4催化剂对低级(富含沥青)的褐煤进行直接煤液化(DCL)。他们首先对合成的Co/SiO2催化剂进行热处理增加金属的分散性,然后采用新型磁性SiO2/Fe3O4作为载体,以便更好地回收催化剂。与褐煤的直接液化结合增加石油产量,进一步提高整体的工艺经济效果。实验表明,富含沥青的褐煤比神华煤更具反应活性,而且在循环使用的过程中,催化剂的活性并未有任何改变。
4 结论与展望
在可持续发展和绿色化学发展的高需求背景下,磁性纳米催化剂不但在活性方面如转化率、选择性等优于传统催化剂,而且极易为外加磁场所分离,同时具有催化活性和催化选择性。将磁性纳米催化剂引入到石油化工产品的生产中,既克服了一般固体催化剂存在反应界面小、传质阻力大等缺点,又解决了均相催化剂难回收、易污染的问题,保证了石油化工产品的绿色生产,简化了操作流程,降低了其生产成本。
虽然磁性纳米催化剂在石油化工产品的生产中已经取得了显著进步,但在许多情况下仍需进一步改进。(1)为了克服目前存在的问题,例如金属纳米颗粒固有的不稳定性和催化剂在苛刻条件下的浸出,仍然需要开发新的多官能团化材料和固定催化剂单元的有效方法。(2)磁性纳米复合粒子的制备步骤较为复杂,大规模应用较难,如何实现磁性纳米催化剂的便捷制备,简化生产工艺,以实现大规模生产应用仍是巨大挑战。(3)目前大部分磁性催化剂的制备过程均使用有机溶剂作为反应介质,从环境友好的角度出发,需要选择环境友好的溶剂体系来制备磁性纳米催化剂,进而避免制备过程中有机溶剂的污染。(4)磁性纳米颗粒在反应过程中易发生团聚与聚沉,如何改善载体表面性质,提高所负载活性基团的稳定性。(5)如何设计新型多功能催化剂允许更多的底物作为起始材料,通过一步反应中的几个步骤生产化学品,从而避免中间体的分离和纯化[79]。(6)如何进一步提高载体表面的吸附量及其磁响应性,以便进一步提高催化活性以及分离效率。(7)有必要对催化剂的转化反应机理和催化剂的结构性质有更深入的了解,从科学和实践的角度来看具有重要意义。尽管这些工艺在实际应用中还有待改进,但我们相信进一步发展将不断研究出更具有使用价值和经济效益的磁性催化剂,并证明该体系的可行性和高产性。相信未来磁性催化剂将会有更多更好的应用以及发展。