



Contents
1 Introduction
2 Extensive application and exposure of BPA
3 Effects of low-dose BPA on human health
4 Key role of transmembrane receptor in low-dose BPA effects
4.1 Effects of low-dose BPA on male germ cells via GPR30
4.2 Effects of low-dose BPA on TH transcription by the αvβ3
5 Conclusion and outlook
1 引言
近年来,环境雌激素对人类健康的潜在危害越来越引起人们关注。这类化合物也称为外源性雌激素,属于典型的环境内分泌干扰物[1],它与生理雌激素分子结构相似,通过模拟或抑制内源性雌激素的生理和生化作用,影响激素受体家族,或干扰内源性激素的产生,继而改变内分泌与生殖系统的正常功能诱发生殖、发育及恶性肿瘤疾病[2]。典型的环境雌激素包括双酚A (Bisphenol A, BPA)、烷基酚和邻苯二甲酸酯等[3]。近年来,BPA在环境相关低剂量下对人体健康的潜在影响受到广泛关注[4,5,6,7,8,9,10]。本课题组在这方面开展了系列工作。本文将结合我们近年来的研究工作,综述目前环境低剂量BPA暴露对人体健康影响的分子机制研究进展、存在的问题以及将来研究的一些思考。
2 BPA的广泛应用及环境暴露
BPA学名为2,2-(4,4'-二羟基联苯)丙烷,系苯酚衍生物,由刚性平面芳环和可塑的非线型脂肪侧链组成。BPA主要作为中间体用于生产聚碳酸酯塑料和环氧树脂等多种高分子材料,广泛用于制造食品和饮料的包装材料(如奶瓶)和内衬、数字媒体(如CDs和DVDs)、汽车、电子设备、运动安全装备和医疗器械(如牙科封闭剂)等[11]。此外,少量的BPA也用于制造酚醛树脂、苯酚类树脂、不饱和聚酯树脂、热敏纸制造的添加剂、火焰阻燃剂(如四溴双酚A)、热和无碳复写纸涂料等[5,8,9]。BPA在不同产品中的大量应用使其全球需求每年以6%~10%的速度增长,特别是在经济快速发展的国家,如中国[11,12,13]。据预测,2011年全球双酚A 产能将达到约560万吨/年,中国总产能将达到61.6万吨/年[14]。电子产品、汽车、建筑和通信装备(计算机和特别是光学媒体,)是中国BPA需求增长的主要驱动力[13]。
通过酯键连接的BPA分子聚合物在高温、酸碱性条件下容易发生水解。普通人群可通过呼吸、皮肤和消化道等途径暴露于BPA[5,9,15,16]。美国疾病预防控制中心的调查研究发现,2500名普通志愿者的尿液样品中有92.6%的样品检测出BPA, 且儿童的BPA暴露水平显著高于成年人(4.5 ng/mL vs 2.5 ng/mL)[17]。此外,在北美洲和多个亚洲国家的普通人群中,尿BPA的检出率高达91%~99%[17,18]。特别是最近的研究显示,BPA在普通人群的血液、唾液、乳腺组织、怀孕妇女的羊水、新生儿血液、胎盘、动脉血和乳汁中也存在[5,19,20]。尽管我国在BPA体内暴露方面的研究比较少,但是,考虑到我国是BPA生产和应用大国,可以预测BPA的环境暴露和人体内暴露具有普遍性[12]。在我国一些地区的河流和地表水以及普通人群,也都不同程度检测到BPA的存在[12]。Wang等[21]发现,职业暴露BPA工人的血清BPA浓度在27.16~41.61 μg/L, 而无暴露的对照组则在0.58~13.54 μg/L。
3 环境相关低剂量BPA对人体健康的潜在影响及分子机制
大量的体内外研究表明,BPA可以对神经、生殖、心血管和能量代谢系统产生潜在影响,引起神经行为异常、生殖功能障碍、心血管功能失常、代谢紊乱等,此外,在乳腺癌、卵巢癌等与雌激素相关的恶性肿瘤的发生和发展中也扮演了关键的角色[22]。到目前为止,BPA对生殖功能、乳腺发育、认知功能和代谢产生的不利影响已被证实并为学界认可,且与人体健康具有密切的相关性[7,23]。BPA对发情周期及动情周期动态变化有负面影响,可以促使周期延长或无规则[7]。来自啮齿动物和体外啮齿动物/人类细胞的数据表明,BPA可以影响卵巢雌激素的产生[24,25,26]。由于雌激素在啮齿动物和人类中具有相似的功能[7],因此推测BPA很可能通过破坏卵巢滤泡活性而影响人体的卵巢周期。最近一项研究表明,BPA可以影响下丘脑神经肽kisseptin和促性腺激素释放激素(GnRH)的释放,导致下丘脑神经内分泌功能改变[27]。Kisspetin也被证明在人体HPG轴控制中发挥作用。因此,BPA被认为可能对人体下丘脑kisspeptin / GnRH系统和动情周期性产生潜在影响[7]。最新的证据表明,BPA可以改变乳腺上皮-间质间的相互作用,产前或产后暴露可增加女性乳腺中末端芽数量、侧支、导管和导管内增生,从而导致对化学致癌物的敏感性增加[5,28,29]。由于人体与啮齿动物乳腺结构相似且细胞增殖受共同的激素调节,推测BPA的早期暴露可能同样诱导人体乳腺的异常发育,从而增加乳腺对致癌污染物的易感性。BPA也被发现与啮齿类动物的学习和记忆行为受损有关,并且能够以性别依赖性方式改变其组织学结构。在非人灵长类动物实验中,已证明BPA产前暴露可对中脑的多巴胺能系统和海马的脊柱突触产生不良影响[30],并且在成年时暴露会产生明显的认知障碍[31]。此外,许多证据表明,BPA还能够影响啮齿动物的行为和脊柱密度[32,33],而对于非人灵长类动物,雌二醇在海马和前额叶皮质中可诱导突触产生[34]。由于性激素或者类固醇类激素对啮齿动物和人类认知的调节作用具有相似性,因此,BPA对啮齿动物学习和记忆的损害被认为也可能在人体中发生。2014年,ANSES (French Agency for Food, Environmental and Occupational Health Safety)发现BPA暴露后能诱导代谢紊乱,引起糖尿病和肥胖[28]。体内证据表明,BPA在胰岛素敏感器官中能够影响β-胰腺细胞的胰岛素合成和释放及胰岛素相关信号的活化,增加2型糖尿病的发病率[7]。同样,大量体外研究显示,BPA通过影响瘦素或脂联素对脂肪细胞分化和功能产生不利影响[7]。此外,产前暴露于BPA可改变机体内葡萄糖的稳态,导致严重的葡萄糖耐受不良和胰岛素抵抗,高胰岛素血症和高瘦素血症。由于在人类和啮齿动物具有类似的胰岛素稳态调节机制及敏感性,BPA对啮齿动物新陈代谢的影响被认为在人体中也可能发生[7]。
BPA在体内外研究中表现出来的雌激素样内分泌干扰效应毋庸置疑[7,35,36]。但是,真实环境中BPA的人体暴露水平很低[20]。Ikezuki等发现,普通人群血清及尿液中非共轭结合的BPA 含量仅在0.1~10 ppb[37]。在“剂量大小决定危害”的传统毒理学观点引导下,有专家认为:日常生活环境中人们接触的BPA浓度很低,不足以对人体健康构成危害。那么,低水平BPA暴露是否确实能对人体健康产生潜在危害?近年来,越来越多的研究发现,BPA可以对实验动物表现出低剂量效应,即在一定的低浓度下,BPA可以诱导明显的毒性效应,如神经、生殖内分泌干扰效应等;而当浓度进一步升高时,这种毒性效应降低甚至完全消失[35,36]。但是,由于低剂量BPA的生物效应难以在人体上复制;BPA对生物体的影响程度,作用范围因种属、暴露剂量、暴露方式的不同也各不相同。此外,也缺乏确切的流行病学证据表明低剂量BPA暴露与某些疾病具有直接联系[4,35,36,38]。因此,目前对低剂量BPA的健康安全问题还存在争议。鉴于BPA在工业生产中的重要地位,同时又考虑到它在低剂量暴露时对人体健康可能的潜在危害,著名毒理学家Borrell在Nature发文呼吁对低剂量BPA的潜在健康危害进行更深入全面的评价,特别是要更关注于其低剂量效应分子机制方面的研究[39]。因为对分子机制的深入理解不仅有助于对体内毒性效应进行预测,而且可以在不同种属之间进行生物效应比较,如关键的信号传导通路,从而能更客观真实地评价和预测环境暴露BPA对人体健康的可能潜在影响以及采取有针对性的预防和干预措施。同时,由于BPA是一种典型的雌激素样内分泌干扰物,对其低剂量效应分子机制的深入探究也将为评价其他类似结构雌激素样环境内分泌干扰物的健康效应提供理论基础及技术支持。国际权威环境与健康医学杂志Environ. Health Perspect. 近年来也一直在持续关注BPA的低剂量效应,刊登了一系列关于BPA低剂量效应机制的论文,并呼吁对低剂量BPA的内分泌干扰机制进行更深入的探讨[40,41,42,43,44,45,46,47]。因此,对低剂量BPA健康效应分子机制的研究不仅具有一定的社会经济意义,而且具有重要的科学意义和实践价值。
4 膜受体介导的非基因组调控方式在BPA低剂量效应中扮演关键角色
体外结合实验表明,大多数雌激素样化合物在环境暴露相关低剂量下与雌激素受体(Estrogen receptors, ERs)结合力很微弱,BPA与ERs结合能力本身就比与雌二醇(Estradiol,E2)的结合能力低1000~10 000倍[48,49]。但是, 在同样的剂量下却可对人和动物产生生物学效应[19,35,50]。由于环境雌激素样化合物通常被认为是通过与雌激素竞争结合ERs而发挥其内分泌干扰作用。因此,在低剂量雌激素样化合物与ERs微弱结合或不能结合的情况下,推测另一种机制可能参与了低剂量雌激素样化合物的内分泌干扰作用。近年来的研究表明,雌激素除了通过ERs调控基因表达外,还能通过膜受体以非基因组调控方式快速激活不依赖于ERs的信号传导通路促使细胞增殖、凋亡[51,52,53]。非基因组调控方式主要通过受体介导的信号传导通路或网络对细胞内重要分子事件进行调控,由于不涉及基因转录和蛋白表达,因此,具有发挥生物效应快、调控精准等特点,为调控许多重要细胞功能所必需。基于雌激素的这种非基因组调控机制,大量体外研究表明,低剂量BPA也可以通过非基因组调控的方式对胰岛、内皮、乳腺和垂体产生快速激活效应[54,55,56]。但是,低剂量BPA通过具体何种膜受体介导这种非基因组调控效应目前还不清楚。膜受体作为BPA可能的直接作用位点以及非基因组调控的起始点,不仅在BPA的低剂量效应机制探讨中具有重要作用,而且为进一步干预这种低剂量非基因组调控效应提供了作用位点和理论依据。因此,对于参与低剂量BPA非基因组调控效应的膜受体鉴定和相关机制研究一直是学界关注的重点,也是迫切需要解决的关键科学问题。近年来,我们在这方面开展了系列工作,发现细胞膜G蛋白偶联受体30 (G protein-coupled receptor 30, GPR30) 和整合素αvβ3及其介导的信号传导通路在BPA的低剂量内分泌干扰效应中扮演了关键的角色。
4.1 环境相关低剂量BPA通过GPR30对雄性生殖细胞功能的影响
如前所述,BPA的低剂量效应被认为是通过膜受体介导的非基因组调控方式所诱导。但具体何种膜受体还不清楚。1998 年,O’Dowd 等在进行快速非基因效应研究时发现了ER-α和β 之外的第3种雌激素受体GPR30[61],该受体与传统雌激素受体没有同源性,为375 个氨基酸残基组成的7次跨膜G 蛋白偶联受体,主要通过第二信使环腺苷酸(Cyclic adenosine monophosphate, cAMP)和表皮生长因子受体(Epidermal growth factor receptor, EGFR)激活后的蛋白激酶途径介导快速非基因效应[62]。GPR30参与了多种生命活动的调节,与能量平衡、脂代谢、心血管功能和神经记忆调节等均有密切关系; 特别是在多种雌激素相关肿瘤的发生及发展中发挥了重要的介导作用[63]。
我们利用鼠精原细胞系GC-1作为实验模型。鼠精原细胞系GC-1具有与B型精原细胞和早期精母细胞共同的特征,这些特征与发育前10 d鼠睾丸主要生殖细胞类型特征相似,减数分裂前生殖细胞中主要为精原细胞[65]。此外,GC-1细胞表达GPR30,有助于观察GPR30在BPA诱导非基因组效应中的作用。
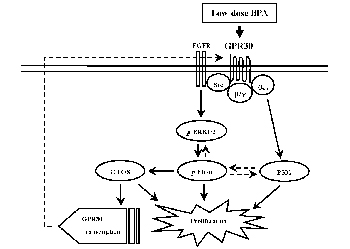
通过细胞存活和增殖实验相互验证,我们发现,BPA 在环境相关低剂量10-11~10-7 M范围内能显著诱导GC-1细胞增殖,其剂量效应曲线呈非基因组调控方式所特有的倒U型 (图1)[45]。在此剂量范围内对细胞凋亡关键执行酶Caspase-3活性没有影响,排除了BPA的增殖诱导可能是通过抑制细胞凋亡的可能性。进一步的研究表明,BPA 对GC-1细胞的增殖诱导主要是通过激活PKG 和EGFR-细胞外信号调节激酶(Extracellular regulated kinase, ERK)信号通路。在此实验中,Gαi/Gαq抑制剂PTX对BPA的细胞增殖效应具有显著的抑制作用,而Gαi/Gαq是细胞膜G蛋白偶联受体(G protein-coupled receptor, GPCR)胞内的重要组成部分,参与了对下游信号通路的激活, 提示BPA对细胞的增殖诱导可能涉及GPCR。在非基因组调控方式中,PKG和ERK信号通路通过快速激活细胞周期调控关键转录因子环腺苷酸效应元件结合蛋白(cAMP responsive element-binding protein, CREB)和母细胞瘤蛋白(Retinoblastoma,Rb)诱导细胞增殖[66]。BPA被发现在15 min时就通过激活ERK和PKG促使 CREB和Rb磷酸化。同时我们也发现,E2-BSA(一种不能跨膜的E2复合物, 通过作用于膜受体发挥其雌激素效应)/BPA 联合与单独给予可以诱导相同的增殖效应,表明这两种物质共存时对GC-1细胞不存在协同或拮抗作用,提示可能通过相似的膜受体及信号传导通路诱导细胞增殖。但是,一个有趣的现象是,BPA和E2联合可以明显促进E2对GC-1 细胞增殖,说明ERs可能参与了BPA 诱导的GC-1 细胞增殖。进一步的数据显示,ER拮抗剂ICI可以显著抑制E2和BPA对GC-1细胞的增殖作用。但是,ICI 对E2-BSA的促细胞增殖作用却没有影响, 表明BPA对GC-1细胞的增殖作用可能同时依赖于ER-α(GC-1细胞不表达ER-β, 且ER-α在膜不表达)和GPCR-EFGR-ERK信号通路。但是,在瞬时转染ER-α报告基因的GC-1细胞和ERs阴性乳腺癌细胞SkBr3中,10-9 M BPA都不能诱导ER-α转录激活。此外,BPA也不能激活转染包含酵母转录因子Gal4DNA结合域和雌激素结合域的ER-α或ER-β。与BPA和E2-BSA对GC-1细胞的联合作用相似,共同给予BPA和GPR30特异性激动剂G1与单独给予诱导相似的增殖效应,提示BPA和G1可能通过相似的信号通路诱导GC-1细胞增殖。但是,与ER-α特异性激动剂PPT(4,4',4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol),BPA 和G1单独作用相比,PPT和BPA或G1联合可以明显促进细胞增殖。此外,利用反义寡核苷酸沉默GPR30或ER-α基因后,可以显著抑制BPA,E2,G1和PPT对GC-1细胞增殖的诱导(图 2)[45]。但是,GPR30基因沉默不能完全抑制E2和PPT对GC-1细胞的增殖作用。证明GPR30参与了BPA的细胞增殖诱导,且GPR30和ER-α可能存在相互调控关系,至今关于GPR30和ERs之间的调控还未见报道,可能具有重要的生物学意义。


我们进一步发现,BPA可以通过ER-α和GPR30介导的EGFR-ERK信号通路显著诱导C-FOS基因和蛋白表达(高达3~8倍)。C-FOS是一种重要的核蛋白转录因子,调控与细胞存活,增殖,侵入,分化相关基因的表达[71],其基因表达的微小变化可以对表型产生明显影响[72,73,74]。一个有趣的现象是,近来的研究表明,通过一个GPR30-EGFR-ERK-C-FOS正反馈循环,C-FOS可以被募集结合到GPR30基因启动子区的激活因子蛋白1(Activator protein-1, AP-1)位点诱导GPR30表达,进而也促使C-FOS表达[75]。BPA对C-FOS的高表达诱导提示在BPA的细胞增殖诱导中可能也存在这样一个正反馈循环。我们发现,BPA确实可以通过ER-α, PKG, EGFR-ERK诱导GPR30基因和蛋白显著表达。通过系列报告基因及染色质免疫共沉淀实验,10-9 M BPA被发现通过相同信号通路募集C-FOS到GRP30基因启动子区(5’区的358碱基片段,包含C-FOS结合的AP-1位点)诱导GPR30转录, 且区内AP-1位点的-210-204碱基为转录激活所必须(图 3)[70]。进一步通过遗传操作使C-FOS失去与AP-1的结合能力后,能显著抑制BPA对GPR30, p-ERK1/2, p-Ser118-ER-α表达的诱导和细胞增殖。值得注意的是,在细胞增殖实验中,C-FOS去功能并不能完全抑制BPA诱导的细胞增殖,提示BPA对GPR30表达的诱导仅仅能促进GC-1细胞进一步增殖,但不能刺激细胞增殖。
研究表明,出生后雄性生殖细胞周期主要由ER-β调控,其下调与睾丸精原细胞癌显著相关[83,84]。因此,在出生前的妊娠期,生殖母细胞(直到出生前不表达活化的ER-β1的异构体)可能特异性对BPA介导的增殖效应特别敏感[85]。因此,胚胎期暴露于BPA可能对雄性生殖细胞造成潜在危险。一组流行病学数据显示,妊娠期母亲和她们的胎儿的血BPA水平分别为0.3~18.9 ng/mL(1.31×10-9 ~ 8.28×10-8 M)和0.2~9.2 ng/mL (8.76×10-10 ~ 4.29×10-8 M)[86]。这些血液BPA浓度与我们目前研究所用相似,提示妊娠期暴露于低剂量BPA可能对胎儿产生不利的生物效应。
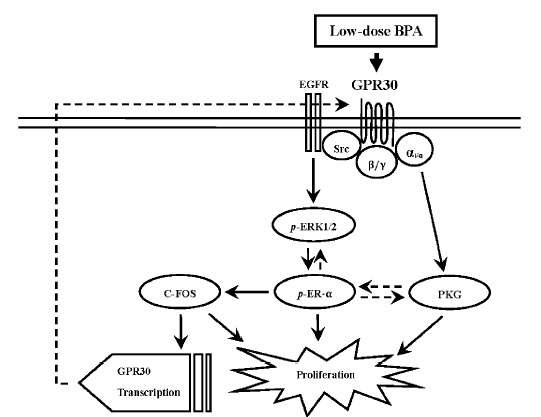
4.2 环境相关低剂量BPA通过膜整合素αvβ3受体抑制甲状腺素调控的转录激活
甲状腺激素(Thyroid hormone, TH),包括三碘甲状腺素(Thiiodothronine, T3)和四碘甲状腺素(Thyroxine, T4),在促进机体基础代谢,特别是大脑生长发育等方面至关重要,它的轻微异常会导致多种认知障碍[87]。因此,能影响TH产生或干扰TH信号通路的环境污染物很可能会对机体发育产生深远影响。了解环境化合物对TH信号通路的影响对人体健康评价至关重要。
TH主要通过与细胞核内甲状腺素受体(Thyroid hormone receptor, TR)形成复合体,促使转录抑制因子核受体共阻遏蛋白(Nuclear receptor co-repressor, N-CoR)/视黄酸和甲状腺素受体沉默中介蛋白(Silencing mediator for retinoid and thyroid hormone receptors, SMRT) 从转录复合体脱落,而募集转录激活因子类固醇受体共激活蛋白-1(Steroid receptor coactivator-1, SRC-1) 到TR的DNA结合域,从而启动基因转录,这种调控模式也称为经典的TH基因组调控模式[88]。BPA与其溴化和氯化衍生物被认为是最早发现可以与TR结合的一类环境化学物质[89]。体外研究表明,双酚A能以相对较低的亲合力结合TR[35],拮抗TH通过TR对基因的转录调控[89,90,91]。体内研究也证实,发育期暴露于BPA可在垂体水平通过选择性拮抗TR-β受体而抑制小鼠的甲状腺素负反馈调节,促使T4水平升高,甲状腺素调控基因蛋白激酶c底物/神经粒蛋白表达增加[92]。BPA与TR的低亲合力意味着需要高水平的BPA才能拮抗TH的转录调控。但是,围产期暴露于低剂量BPA的大鼠,BPA也会通过TH通路影响其子代的神经发育,在出生后15天具有较高的T4水平,脑部TH调控基因上调,导致脑部出现认知障碍,高反应性等发育异常现象[92]。Iwamuro等的研究也表明,低剂量BPA(0.1 μM)可以明显拮抗非洲爪蟾蝌蚪尾部TR的基因表达,特别是当与TH共暴露时,BPA的拮抗作用更加明显[90]。Nakamura等发现,妊娠期BPA低剂量暴露(20 μg/kg/d)会导致子代大鼠大脑皮层组织发生变化和TR-α及其相关基因的表达[93]。围产期暴露于低剂量BPA被认为通过一种非基因组调控的方式影响甲状腺素功能和脑发育,因为TR不是一个直接作用靶点[94],但是,低剂量BPA干扰TH效应的具体机制还不清楚。
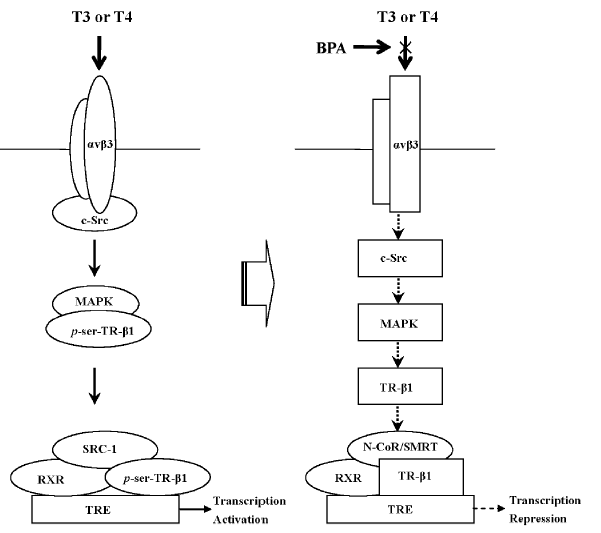
TH的转录调控作用在不同类型的细胞中并不完全通过TR,有时通过非基因组方式,也就是通过快速激活膜受体介导的信号通路而发挥其调控功能,TR的非基因组作用方式至少部分依赖于整合素αvβ3和由其调控的细胞信号通路[95]。整合素αvβ3是由αv亚基和β3亚基通过1:1比例以非共价键结合而成的异二聚体跨膜糖蛋白,属于黏附分子家族重要成员。每种亚基都由胞质外区、跨膜区和胞质内区三个部分组成。胞质外区与特异性配体结合,胞质内区通过与肌动蛋白结合蛋白,细胞内信号蛋白和非酶类蛋白等结合,调控细胞之间,细胞与细胞外基质之间的通讯(如细胞伸展、迁移和细胞外基质的装配),以及细胞内外信号的双向传导(如粘着斑激酶调控的MAPK信号通路等)[96]。T3和T4是αvβ3的激动剂[98],通过激活αvβ3介导的MAPK/c-Src信号通路使核内TR-β DNA结合域(TR-β DNA binding domain, TR-β-DBD)142位的丝氨酸磷酸化,促使N-CoR或SMRT从TR分离,激活基因转录。
大量研究表明,低剂量BPA主要通过直接调控膜受体介导的细胞内信号传导通路,以非基因组调控的方式发挥其内分泌干扰效应[35]。我们推测,环境相关低剂量BPA的TH干扰效应也可能是通过直接作用于整合素αvβ3介导的相关信号通路而拮抗T3/T4调控的转录激活。我们利用非洲绿猴肾细胞系CV-1作为实验模型,这种细胞便于遗传操作,且缺乏TR,但表达αvβ3,便于研究αvβ3及其介导信号通路在BPA对TH/TR转录拮抗中的作用。
基因实验结果显示,BPA 在10-9~10-7 M 浓度范围内显著抑制生理浓度T3介导的TR-β1 转录。在此剂量范围内对细胞无毒性效应,排除了BPA的转录抑制效应是由于细胞存活率下降所致的可能性。BPA 不能抑制维甲酸X受体和糖皮质激素受体介导的转录,表明BPA 对TR 转录抑制具有特异性。进一步,我们也发现,在缺乏T3的条件下,BPA在此低剂量范围也不会影响本底转录和TR-β1介导的基本转录,以及N-CoR介导的TR-β1转录抑制,表明BPA对TR转录的抑制不是通过与TR直接结合。确实,目前研究所用的BPA浓度与TR的亲合力远远低于T3[91]。以上这些结果表明,低剂量BPA对TR转录的抑制效应可能不是通过与TH竞争结合TR所致。利用哺乳细胞双杂交和谷胱甘肽巯基转移酶下拉实验,我们发现,当共转染转录激活因子SRC-1时,BPA不能直接影响T3介导的TR-β1与SRC-1的结合,排除了BPA直接影响TR-β1与SRC-1结合进而抑制转录的可能性。当与生理浓度T3/T4共暴露时,10-9~10-7M BPA可以剂量依赖地促使转录抑制因子N-CoR/SMRT与TR-β1结合。 Moriyama等发现,高剂量BPA通过直接作用于TR也可以募集N-CoR到TR,拮抗TH的转录调控[91]。有趣的是,他们的数据显示,BPA对T3结合TR的抑制常数约为10-4 M,但是,10-6 M BPA已经显著抑制TR转录。进一步,10-9~10-5 M BPA不能明显结合TR,但是能显著募集N-CoR到TR。综合已有结果,提示低剂量BPA可能通过其他方式募集N-CoR到TR。
那么,是否整合素αvβ3及其调控的c-Src信号通路介导了低剂量BPA对N-CoR的募集?我们发现,在T3/T4 存在的情况下,αvβ3 整合素和c-Src 过表达可以明显抑制BPA 对N-CoR/SMRT 的募集,以及相应的TR转录抑制效应(图 5)[99]。证明β3 整合素和信号蛋白c-Src 参与了BPA 对N-CoR/SMRT 的募集和对TR-β1的转录抑制。T3可以结合αvβ3的两个位点,激活MAPK和/或c-Src/PI3K通路,而T4仅仅结合一个位点激活MAPK[98]。但是,我们发现,c-Src也涉及到BPA诱导的T4调控的N-CoR或SMRT到TR-β1的募集,提示BPA通过αvβ3作用后,在细胞内PI3K和MAPK之间有相互作用。这些发现可能有其他重要的生物学意义。
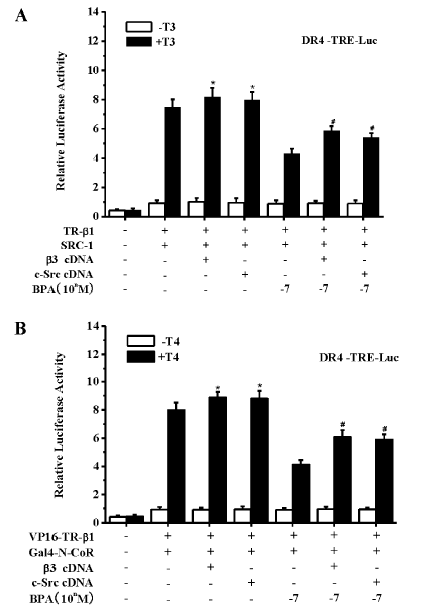
Bhargava等的研究表明,TH通过激活αvβ3/c-Src/MAPK信号通路诱导使TR-β1的丝氨酸磷酸化,导致N-CoR或SMRT从TR-β1解离,激活转录[98]。因此,我们进一步确证了BPA是否通过干扰TH激活的αvβ3/c-Src/MAPK/丝氨酸磷酸化的TR-β1通路而拮抗了其转录激活。在缺乏TR的293T细胞,免疫共沉淀实验结果显示,生理浓度T3或T4能明显抑制TR-β1与N-CoR或SMRT形成共沉淀。但是当与BPA, MAPK抑制剂PD98059, c-Src抑制剂PP2, 以及αvβ3整合素拮抗剂Tetrac共暴露时,T3/T4调控的这种TR-β1/N-CoR或SMRT共沉淀抑制效应能被明显拮抗。证明整合素αvβ3以及其调控的信号蛋白c-Src和MAPK参与了BPA对TH/TR转录激活的拮抗。进一步,在T3/T4存在时,BPA,PD98059,PP2或Tetrac也可以显著抑制TR-β1与酪氨酸磷酸化的MAPK或丝氨酸磷酸化的TR-β1形成共沉淀,表明BPA抑制了TH对MAPK和TR-β1的激活。此外,BPA, PP2和Tetrac 被发现可以抑制TH介导的αvβ3与c-Src免疫共沉淀的形成,但是PD98059则不能。证明BPA是通过αvβ3/c-Src通路拮抗了MAPK对TR-β1的丝氨酸磷酸化。以上结果表明,BPA通过干扰TH调控的αvβ3/c-Src/MAPK /TR-β1通路而抑制其转录激活,同时也进一步确证了TH通过此通路促使N-CoR/SMRT从TR-β1解离,利于进一步转录激活。
妊娠期大鼠饮食暴露于低剂量BPA可使子代血清T4水平升高,脑部蛋白激酶c底物/神经粒蛋白表达改变[92]。在这个研究中,BPA剂量和血清T4水平之间的“平缓”的剂量效应关系被认为是由于有限量的TR和共抑制因子所致。但是,基于我们目前的结果,推测限量的αvβ3和TR-β1可能部分有助于BPA对血清T4水平的“平缓效应”。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论及进一步亟待解决的关键问题
GPR30是新发现的一种重要雌激素膜表面受体,在人和鼠之间高度保守,广泛表达于不同组织细胞,在神经、生殖内分泌系统等介导多种生物学效应[62,95]。整合素受体αvβ3为细胞黏附分子家族的重要成员之一,表达于多种组织细胞,在不同种属间高度保守[96]。T3/T4通过αvβ3介导的非基因组调控参与了体内多种生理病理过程[95]。基于此,我们推测低剂量BPA对膜受体GPR30或/和αvβ3介导非基因组效应的影响可能在诱发体内内分泌干扰效应中扮演了关键的角色。此外,由于它们的蛋白结构功能与人高度保守,提示低剂量BPA也可能通过干扰这两种受体介导的非基因组调控效应对人体正常生理功能产生影响。在我们的研究中,一个有趣的现象是,GPR30介导的ERK信号通路可以使ER-α分子的118位丝氨酸磷酸化而使其持续激活,同样,αvβ3整合素介导的ERK信号通路也以同样的激活方式使ER-α活化。因此,我们推测,在体内环境(T3/T4同时存在)时,GPR30和αvβ3之间可能经由不同信号传导通路相互协调配合而共同介导BPA的低剂量效应。因此,在大量表达GPR30和αvβ3的神经和生殖细胞中,环境相关低剂量BPA通过GPR30或/和αvβ3调控了哪些信号传导通路以及相应的影响了哪些细胞功能,这是我们需要进一步探讨的第一个关键问题。
近年来,环境内分泌干扰物对表观遗传的影响受到广泛关注[100]。表观遗传主要是指不涉及DNA序列改变的基因表达和调控的可遗传修饰,它是环境因素与细胞的遗传物质交互作用的结果,因而它参与了许多疾病的发生和发展,特别是肿瘤[101]。DNA甲基化作为表观遗传修饰的一种重要方式,是研究最为充分的一种表观遗传学事件,也是目前环境内分泌干扰物对表观遗传影响的主要作用方式[102]。DNA甲基化是指在DNA甲基转移酶或/和甲基结合蛋白作用下,以S-腺苷甲硫氨酸为甲基供体,将甲基基团转移到胞嘧啶和鸟嘌呤二核苷酸的胞嘧啶。DNA甲基化能引起染色质结构、DNA构象、DNA稳定性及DNA与蛋白质相互作用方式的改变,从而控制基因表达,在细胞正常发育、基因表达模式以及基因组稳定性中起着至关重要的作用。研究表明,环境雌激素样内分泌干扰物通过ERK信号通路介导的ER-α 功能区AF-1磷酸化对生长因子的正反馈调控可能与DNA甲基化修饰有关[103]。我们的研究表明,环境相关低剂量BPA通过GPR30-ERK对ER-α AF-1区的激活与表皮生长因子也存在正反馈调控[70]。此外,最近的研究也表明,低剂量BPA可以影响调控细胞基因组DNA甲基化的关键基因和酶[104,105,106]。鉴于此,我们推测,在BPA的低剂量效应中,膜受体GPR30或/和αvβ3介导的非基因组调控可能与表观遗传中的DNA甲基化修饰存在着某种相互调控关系。基于以上分析,将要探讨的第二个问题是:环境相关低剂量BPA如何通过GPR30或/和αvβ3介导的信号传导通路调控DNA甲基化修饰。目前认为,环境内分泌干扰物对个体表观遗传影响的一种主要方式是生殖细胞系依赖的表观遗传调控[107]。环境内分泌干扰物通过影响DNA甲基化对生殖细胞系(精原,卵母细胞等)的遗传发育程序信息进行重新编程。重新编程的遗传信息最可能经过生殖细胞系传递给子代,从而对子代产生各种健康危害[108,109]。因此,如果能明确低剂量BPA影响DNA甲基化修饰的分子机制,将对发育期暴露BPA对子代遗传影响的评价具有重要参考价值。
在体内,环境内分泌干扰物主要是通过干扰内源性激素的生理功能,如拮抗、协同等而发挥其内分泌干扰作用。因此,在体外,当其与体内生理浓度激素共存时可能会产生与其单独暴露不同的生物效应,特别是在低剂量暴露情况下。在我们的低剂量BPA对甲状腺素非基因组效应干扰研究中,BPA单独对细胞没有任何影响(由于此细胞不表达GPR30),但是,当模拟体内真实暴露,让其与生理浓度甲状腺素T3或T4共存时,BPA可以干扰T3/T4对αvβ3的激活[99]。此外,生理浓度雌激素与BPA共存可以改变鼠脑垂体细胞关键信号分子ERK1/2磷酸化状态,但是单独BPA处理却不会产生这种效应[110]。用BPA处理摘除卵巢的大鼠,可以剂量依赖地抑制雌激素诱导的海马CAI区锥体神经元细胞内树突棘突触的形成。在发育中的小脑神经元内,BPA和雌激素共注射可以剂量依赖地抑制雌激素诱导的ERK1/2快速活化。通过分析我们目前的结果,我们认为,上述研究所表明的低剂量BPA对雌激素ERK1/2活化的抑制可能是由于体内BPA与T3/T4和雌激素共暴露所诱导的复合效应所致:雌激素通过膜表面受体激活ERK1/2,同时,由于低剂量BPA通过GPR30激活了相同的信号通路,受饱和激活机制的调控,BPA不会再使ERK1/2更大程度活化,但是它可以干扰T3/T4通过αvβ3对ERK1/2的激活,最后综合作用的结果是对ERK1/2信号通路的抑制。因此,我们推测,目前在体内实验中观察到的BPA低剂量内分泌干扰效应可能是BPA与体内生理浓度激素复合作用的结果。研究表明,雌激素、甲状腺素以及雄激素对于神经、生殖发育调控发挥关键作用,而且,BPA也主要是干扰这几种激素[37]。基于以上分析,我们需要探讨的第三个问题是:在与生理浓度的雌激素、甲状腺素以及雄性激素共存时,环境相关低剂量BPA又是通过GPR30或/和αvβ3具体介导了哪些信号传导通路以及相应的影响了哪些细胞功能及DNA甲基化修饰。如果能了解雌激素、甲状腺素以及雄性激素在生理浓度下与环境相关低剂量BPA的复合暴露效应及机制,将有助于更真实,准确理解低剂量BPA的内分泌干扰效应,从而能对整体动物实验结果以及对人体健康影响进行更合理解释和可靠推测。