
科学观察, 2015, 10(5): 1-14


Cite this article:
Li Zhenqi , Su Yan , Xu Li , Wang Yue , Xu Ping , Yu Jianrong . International Development Trend of Non-Coding RNA . SCIENCE FOCUS [J], 2015, 10(5): 1-14 doi:10.15978/j.cnki.1673-5668.201505001
摘要:
进入21世纪以来,随着人类基因组计划的完成,非编码核糖核酸研究逐渐成为生命科学领域的研究热点。该文以Web of Science数据库为数据源,以Thomson Data Analyzer软件为工具,通过对非编码RNA相关文献进行分析,梳理该学科的发展历程与重点方向,为中国在该领域的发展提供参考。
关键词:
非编码RNA
;
文献计量学
;
长非编码RNA
;
微RNA
;
发展态势
Key words:
non-coding RNA
;
bibliometrics
;
long non-coding RNA
;
microRNA
;
development trend
1 引言
非编码RNA(Non-coding RNA,ncRNA)是指不编码蛋白质的RNA,从长度上可以分为小于50 nt1(1 nt即核苷酸(nucleotide)。)、50 nt~500 nt、大于500 nt三种类型。狭义上的非编码RNA主要是指不包括信使RNA(message RNA,mRNA)、转运RNA(transfer RNA,tRNA)和核糖体RNA(ribosomal RNA,rRNA)的其他RNA分子。而广义上的非编码RNA还包括细胞中含量最高的、获得较为透彻研究的两种常见ncRNA——rRNA与tRNA。
非编码RNA的研究始于最初的rRNA、tRNA、小核RNA(small nuclear RNA,snRNA)和小核仁RNA(small nucleolar RNA,snoRNA),逐渐发展到后来的微RNA(microRNA,miRNA)、小干扰RNA(small interfering RNA,siRNA)及与Piwi蛋白相互作用的RNA(Piwi-interacting RNA,piRNA),再到长链非编码RNA(long non-coding RNA,lncRNA)与环状RNA(circular RNA,circRNA)等。这些种类繁多、长短各异、功能多样的非编码 RNA被认为是基因组的“暗物质”[1],对其最终的认识和理解将对整个生命科学的发展产生难以估量的影响。
自1990年以来,随着基因组研究的不断开展与测序能力的持续提升,海量而又繁杂的基因组序列数据提示我们,编码蛋白质的DNA区域在人类基因组中的比例少于3%,而非编码序列虽然不能够编译蛋白质与多肽,但能够以非编码RNA的形式进行表达[2]。这些发现引起了研究人员的关注,相关研究发展迅速,尤其近10年,非编码RNA研究取得了一系列突破性成果,已经成为生命科学领域的热点之一。自2000年起,非编码RNA的相关研究内容连续多次入选Science杂志年度10大科学突破:2000年的“地球上的生命可能起源于RNA”、2001年的基因沉默和RNA干扰(RNA interference,RNAi)、2002年的小RNA(small RNA)与RNAi的研究成果连续列入当年Science 10大突破;2003年,科学家从早期的基因表达到发育过程进一步探索小RNA对细胞行为的影响;2004年,研究人员证实基因组中的所谓“垃圾DNA”作用要比原先认为的更重要,而这些“垃圾DNA”的产物便是非编码RNA;2006年,科学家们发现一类新的非编码RNA分子piRNA能够与Piwi蛋白家族成员相结合,参与生殖细胞生长发育过程中的调控;2012年,耗时9年的ENCODE项目研究成果表明,人类对于非编码RNA调控基因功能网络产生进一步认识;2013年,CRISPR成为炙手可热的基因组编辑技术,而发挥RNA介导的DNA切割作用所必不可少的辅助因子——CRISPR RNA(crRNA)与反式激活嵌合RNA(trans-activating chimeric RNA,tracrRNA)——均为非编码RNA。此外,两名美国科学家Andrew Z Fire和Craig Mello因证实了siRNA所引起的RNA干扰机制而荣获2006年度的诺贝尔生理学或医学奖[3]。
研究已经表明,非编码 RNA 发挥了非常重要的生物学功能,参与了胚胎发育、干细胞维持、细胞分化、代谢、信号转导、免疫应答、癌症、衰老等几乎所有生理或病理过程的基因表达调控[4]。非编码RNA也与重大疾病(如癌症、心血管疾病)、神经退行性疾病(如阿尔茨海默病、帕金森病)和慢性病(如糖尿病、高血压)等疾病的发生有关,很多非编码RNA可作为药物治疗的潜在靶点。除此之外,由非编码RNA介导的RNAi技术与基因组编辑技术的“金剪刀”CRISPR能够从基因沉默和基因组改造的角度对生物医学的发展作出重大贡献。非编码RNA研究既是生命科学的重要基础前沿,也是促进技术开发和实际应用的典型范例。
2 数据来源与分析方法
文献部分利用Web of Science数据库,以所有已知的非编码RNA种类名称及其缩写形式作为关键词,检索SCI收录的生命科学相关学科分类文章,检索日期为2015年5月18日,文献类型选择Article和Review。采用此途径共检索获得相关文献115 935篇,2005–2014年文献89 487篇。
利用Thomson Data Analyzer(TDA)软件对检索的文献进行字段清洗与内容梳理,对年度、国家/地区、机构、关键词与被引频次等主要指标进行统计分析。利用CiteSpace软件,通过考察词频,将某段时间内频次变化率高的突发词(burst term)从近年来大量的主题词中探测出来,进行引用与聚类分析,形成时间轴与词云(word cloud),用以说明学科的发展历程及前沿热点。
3 非编码RNA学科发展态势
从非编码RNA的论文发表数量上来看(图1),其学科发展大体分为3个阶段:萌芽期、发展期、快速增长期。1966–1990年,该领域的发文数量相对较少;1990年之后,随着人类基因组计划的提出,非编码RNA逐渐引起人们的重视,发文数量稳步攀升;自2001年人类基因组计划草图完成以及非编码RNA在基因调控、基因沉默等生物学机制中重要作用的凸显,相关领域的科研进展与学术研究进入快速增长期。
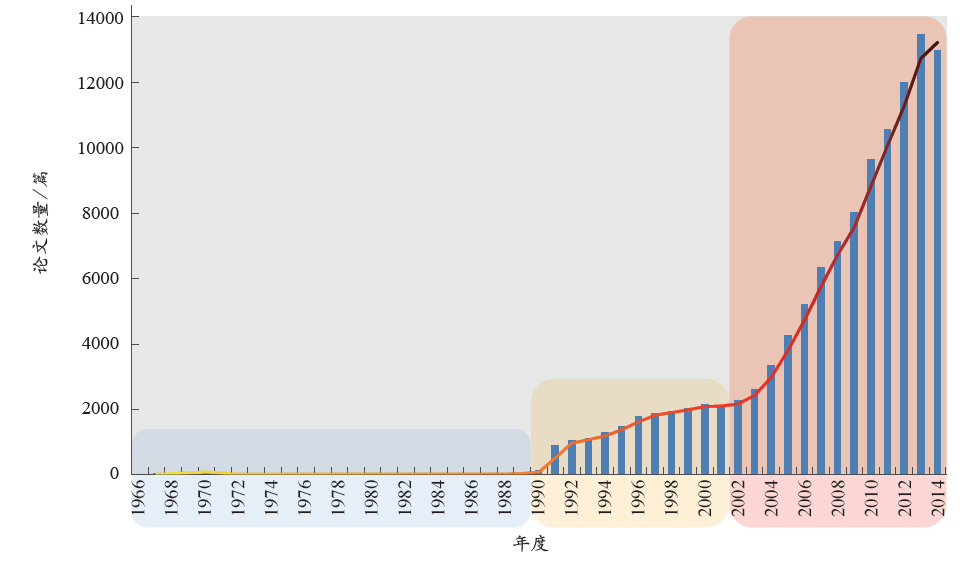
3.1 国际发展态势
(1)第一阶段——萌芽
事实证明,RNA的研究浪潮往往是由DNA的某个重大突破所引起的[5]。20世纪50年代,rRNA和tRNA作为最早的非编码RNA为人们所发现。此次发现并非偶然,正是由于1953年DNA分子双螺旋结构的解析引起了人们在转录和翻译水平对遗传信息的进一步解读,从而促进了这两类非编码RNA的发现。1977年,断裂基因的发现[6]让人们认识到,在基因组水平的遗传信息编码并非是连续性的。随着snRNA的发现[7,8],从RNA转录后加工水平来解读遗传信息表达的过程及机制日渐明晰。1982年,Cech等研究原生动物四膜虫的rRNA时,首次发现rRNA基因转录产物的I型内含子剪切和外显子拼接过程可在无任何蛋白质存在的情况下发生[9],该研究预示细胞中存在大量具有催化功能的调控RNA。起初,人们认为基因研究中最重要的对象是DNA和蛋白质,而RNA只起到传送DNA信息的作用,但RNA编辑现象的发现颠覆了这种认知。1986年,Benne等在锥虫动质体线粒体中发现了RNA编辑现象[10],自此打破了基因与蛋白质的线性传递规则,从而进一步证实非编码RNA能够调控遗传信息的表达。
(2)第二阶段——发展
1990年,该领域的相关论文仅118篇,而1991年爆发式地增长至881篇,并在此数量级上维持了10年左右的稳健发展。追本溯源,这是由于1990年人类基因组计划“横空出世”,揭开了人类基因组序列的神秘面纱,从而带动了该领域的进一步发展。1990年代,通过其对人类基因组序列的测定与分析,科研人员在细胞中陆续发现各种新的非编码RNA。细胞中大量非编码RNA的发现有力地证明非编码RNA结构与功能的多样性及复杂度,宣告了RNA组学新时代的到来。在这个阶段,snoRNA、miRNA与siRNA相关论文的产出比较突出。
1990年代初期,科研人员在真核生物及古细菌中发现大量的snoRNA,构成了不断扩大的“snoRNA世界”[11]。这类小型的非编码RNA分子主要包括C/D box、 H/ACA box、复合H/ACA与C/D box以及孤儿snoRNA等类别,能够引导rRNA或其他RNA的化学修饰(如甲基化)作用[12]。
1993年,Lee等在秀丽隐杆线虫(Caenorhabditis elegan)中发现了第一个能时序调控胚胎后期发育的基因lin-4[13]。时隔7 年,Reinhart 等在秀丽隐杆线虫中又发现了一个异时性开关基因let-7[14],并将这类基因所编码的能时序调控发育进程的小分子RNA称之为时序调节小RNA(small temporal RNA,stRNA)。随着技术的进步,越来越多的此类小RNA在多个物种中被发现。2001年,Science刊文报道在线虫、果蝇和人的cDNA文库中鉴定出近百个与上述发现类似的小分子RNA,并将其统一命名为microRNA[15,16,17]。这也是RNA领域研究的重要里程碑事件。miRNA通过与目标mRNA结合,进而抑制转录后的基因表达,在调控基因表达、细胞周期、生物体发育时序、疾病发生发展等方面起重要作用,具有极其重要的生物学功能与意义。
RNA干扰现象是1990年由Jorgensen研究小组在研究查尔酮合成酶对花青素合成速度的影响时发现的[18]。1992年,Romano和Macino在粗糙链孢霉中发现这样一个事实——外源导入基因能够抑制具有同源序列的内源基因的表达[19]。1995年,Guo和Kemphues在线虫中也发现了RNA干扰现象[20]。经过上述研究的铺垫,Fire等于1998年在Caenorhabditis elegan中发现,加入siRNA能够产生比正义或反义RNA更强的基因表达抑制效果,并将这种现象正式命名为RNAi[3]。由于RNAi在基因沉默方面的简易高效,所以成为了基因功能研究的重要工具,并在药物靶标发现、确认以及疾病治疗方面获得了广泛应用。
1990–2001年,正是由于上述非编码RNA及其相关生物学功能的不断发现再次燃起了人们对“RNA世界”的向往与关注,该领域的文章数量自此得以稳步增长。
(3)第三阶段——快速增长
2001年,人类基因组计划的基本完成宣告了后基因组时代的开端,科研人员即将从非编码RNA的角度对遗传信息进行解读,展开功能基因组学的研究。这预示着又一轮RNA研究高潮即将到来,而事实也恰恰如此。自2002年起,该领域文献的年均增长幅度达到了17.08%;受miRNA与RNAi进入2002年Science杂志10大科学突破的影响,2003–2007年该领域论文数量的年增长幅度均在15%以上,其中后4年增长幅度至少为22%;当ENCODE计划于2012年9月发布其当前完成阶段的成果之后,2013年相关论文涨幅再次达到20%。
2006年7月,Aravin等发现了piRNA的存在[21]。随后,Girard等也检测到了这种非编码RNA[22],并发现它们与生殖细胞发育密切相关。诸多研究表明,数以百万计的piRNA序列存在于生殖细胞之中,其数目远远超过其他非编码RNA总和。因此,piRNA肩负着在生殖细胞发育中调控基因表达的重要任务。它可以与Piwi蛋白结合形成piRNA复合物(piRNA complexes,piRCs),具备沉默转录基因过程、维持生殖系和干细胞功能、调节mRNA的稳定性等生物学功能。
与此同时,在小RNA研究的启示与新型技术的助力下,lncRNA也由于其与人类疾病具有密切联系,逐渐引起了人们的浓厚兴趣。尽管具有基因特异性调控作用的lncRNA(如H19和Xist)在1990年代早期就已经被发现,但随着2005年“转录噪声”观点的普及[23],lncRNA的研究才逐渐引起人们的重视。1990年,Brannan等在哺乳动物的细胞中鉴定出首个lncRNA(H19),并发现其与癌症及胎儿生长有关[24]。随后,Brockdorff等也发现Xist能够关闭第二个雌性X染色体,以确保基因的正确活性[25]。此后,多种lncRNA如雨后春笋般不断涌现在人们面前。它们主要分为[26]:(i)相对独立的不与编码基因重叠的 RNA,如MALAT1[27]和HOTAIR[28];(ii)天然反义转录本,如Xist和Tsix共同控制 X 染色体的失活[29];(iii)假基因;(iv)长的内含子区非编码 RNA,如 COLDAIR[30];(v)与启动子联系的转录本或增强子 RNA,如 pasRNAs[31]和 eRNAs[32]。lncRNA不仅能够调控基因转录及表达、调控基础转录元件,还可以参与转录后的剪接调控、翻译调控以及基因调控,此外还在表观遗传调控中起着重要作用。随着研究的不断深入,科研人员发现大多数lncRNA在癌症和其他重大疾病中的表达,因此它具备作为诊疗生物标记物和药物靶点的巨大潜力,这带动了lncRNA研究热潮的迅速兴起。
近年来颇受关注的circRNA的研究历程与lncRNA类似——发现时间相对较早,相关研究主要集中在最近几年。circRNA是一类不具有5'末端帽子和3'末端poly(A)尾巴、并以共价键形成环形结构的非编码RNA分子[33]。自1976年起,研究人员曾先后在病毒[34,35]和真菌[36]中发现它们的存在。但在此后的数十年中,由于其表达丰度较低,circRNA仅在个别基因中被鉴定出来。随着技术的发展,研究人员先后在多种模式生物、哺乳动物和人类自身发现大量circRNA的存在[37]。近年来,通过新一代的高通量测序技术与专门针对circRNA的实验计算方法相融合,大量circRNA分子在不同物种中被发现[38,39],并识别出其充当miRNA分子海绵、参与基因转录调控、与RNA结合蛋白相互作用以及翻译蛋白质等作用。
3.2 我国发展情况
我国对于RNA的研究曾经一度处于全球领先水平。1960年代,国际生物学领域刚刚开始进行核糖核酸的结构解析和功能理解,我国科学家就逐步开始跟进RNA相关研究。随着在RNA领域研究的不断深入,我国于1981年出色地完成了酵母丙氨酸tRNA的全人工合成,并且在其具有生物活性、能够进行全部碱基修饰的基础上,获得了全球最高的产率与活性。
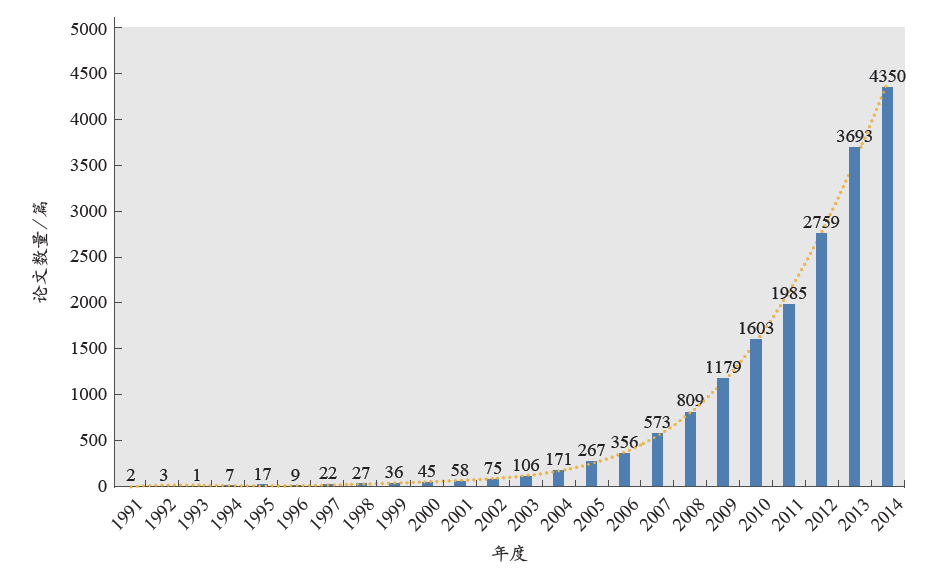
从发文数量上来看,我国非编码RNA学科的起步相对较晚,当全球处于稳健发展期(1990年左右)的时候,我国的ncRNA研究刚刚起步(图2)。虽然我国的学科发展步伐略有滞后,但增长速度令人欣喜。从1998年开始至2002年,论文数量开始稳步攀升。2003年以来,我国的非编码RNA相关文章数量增长迅速,2003–2014年的平均增长幅度为40.87%,年复合增长率达40.17%。这与科学技术部及国家自然科学基金委员会等机构的大力资助有着密切的关系。
3.3 国家/机构水平

从全球在该领域的发文数量分布(表1)可以看出,由于美国在非编码RNA领域的研究起步较早,且资助力度很大,因此占据领军地位,论文产出远超其他各国。最近10年我国在发文数量上为美国的一半左右,排名第二位。根据近10年的数据变化,可以发现虽然英国与法国等RNA研究传统强国起步较早,但近年来已经被韩国赶超。这与韩国的相关政策倾斜密不可分,如韩国先后开展了基因组分析项目、国家生物样本库项目与后基因组计划等大型项目。日本近10年发表的非编码RNA领域的文章数量已经超过德国,同样与哺乳动物基因组功能注释(Functional Annotation of the Mammalian genome,FANTOM)等大型计划的开展有关。
表1
全球ncRNA相关论文数量排名前10国家
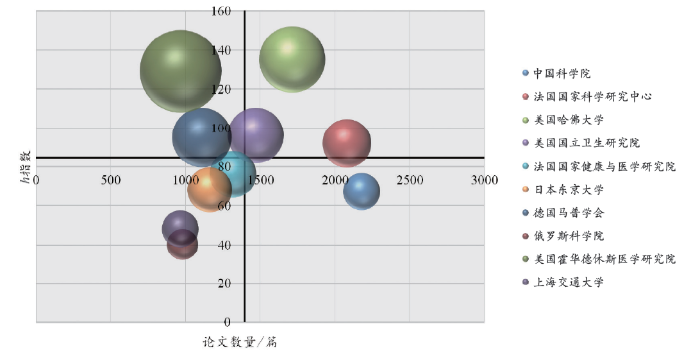
综合论文的发表数量、篇均被引频次和h指数2(2 h指数是指每篇论文至少被引了h次的h篇文章。)能够衡量不同机构的科研竞争力与学术影响力。综观近10年的学术影响力指标(图3)可以发现,中国科学院在发文数量(2 181篇)上已经跃升至全球首位,上海交通大学也出现在非编码RNA研究领域的TOP10机构榜单之上。哈佛大学近10年的高质量研究较多,h指数达到135。在发文数量上处于同一数量级的俄罗斯科学院(981篇)、霍华德休斯医学研究院(969篇)和上海交通大学(966篇)在文章影响力上有很大差异,霍华德休斯医学研究院的篇均被引频次高达69.45,而俄罗斯科学院和上海交通大学此项指标分别为10.04和13.69。
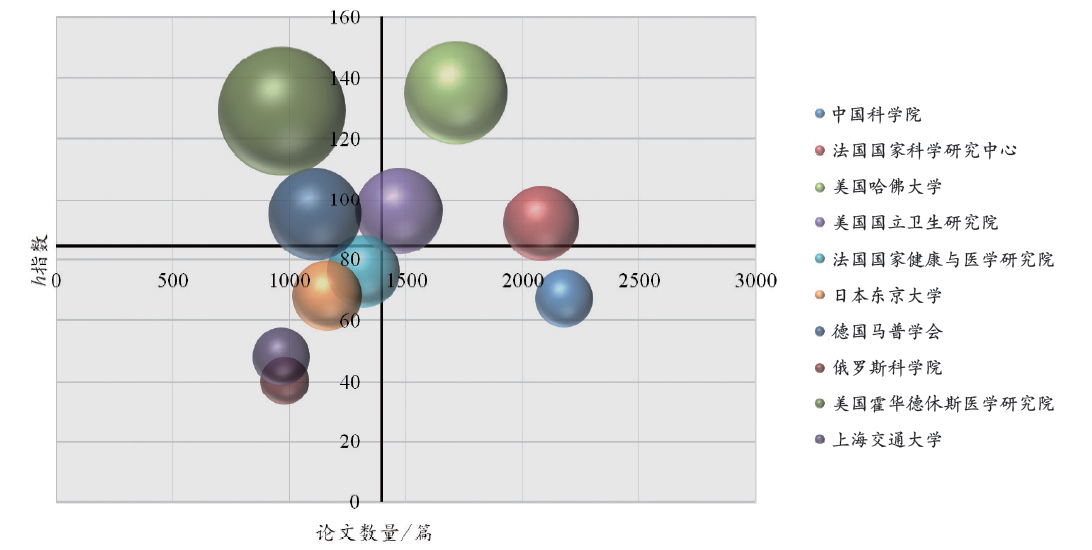
图3
2005–2014年全球ncRNA相关论文数量排名前10机构的学术情况对比
注:气泡越大,说明机构的篇均被引频次越高;两坐标轴交点为前10机构的论文数量与h指数的平均值,因此气泡越靠近右上角说明机构的综合学术影响力越强。
注:气泡越大,说明机构的篇均被引频次越高;两坐标轴交点为前10机构的论文数量与h指数的平均值,因此气泡越靠近右上角说明机构的综合学术影响力越强。
自我国开展ncRNA研究以来,综合考量国内研究机构的学术影响力(表2)能够看出,中国科学院的发文数量(2 414篇)领先于其他国内机构,h指数(72)同样排名首位。这说明中国科学院不仅发文数量较多,而且其中不乏高影响力的论文。香港大学的发文数量(425篇)虽然仅有中国科学院的五分之一左右,但h指数(48)和篇均被引频次(22.09)分别列第2名和第1名,说明香港大学在此领域的学术影响力相对较高。中山大学(h指数53,篇均被引频次15.81)、复旦大学(h指数44,篇均被引频次14.14)和中国医学科学院(h指数40,篇均被引频次15.53)在学术影响力方面表现突出。
表2
1991–2014年中国ncRNA相关论文数量排名前10机构 (以论文数量排序)
3.4 学科前沿热点

对2012–2014年的关键词进行频次与共现关系统计分析(图4),可以看到在非编码RNA研究领域,近3年出现频率比较高的是小RNA、小非编码RNA、特异miRNA、长非编码RNA、转录因子、干细胞、靶基因和癌细胞等研究对象;科研人员关注信号通路、调控网络、RNA干扰、生物学过程、表观遗传机制、DNA甲基化、转录后调控和miRNA表达等相关的基础研究;而从转化医学和临床角度出发,研究聚焦于治疗靶点、临床特征、癌症疗法、人类疾病、发病过程和早期诊断等研究方向。

图4
2012–2014年ncRNA研究领域热点词频率分布
注:图中每个方形代表一个领域热点词,使用文字进行标注。其中图形和文字大小代表词频的高低;每个图形分别计算2012、2013和2014年出现的频率,并使用渐变色予以区分年度变化;连线代表词语之间存在共现关系。
注:图中每个方形代表一个领域热点词,使用文字进行标注。其中图形和文字大小代表词频的高低;每个图形分别计算2012、2013和2014年出现的频率,并使用渐变色予以区分年度变化;连线代表词语之间存在共现关系。
根据该领域热点词的频率分布,发现非编码RNA 研究热点大致可以分为以下几类:(i)非编码RNA及其相关基因的识别与鉴定,如RNA基因(rna genes)、微RNA基因(mirna genes)与微RNA表达(mirna expression)等;(ii)非编码RNA的结构与功能,如生物学功能(biological function)、功能作用(function role)和生物学过程(biological process)等;(iii)非编码RNA的表观遗传调控,如DNA甲基化(dna methylation)、表观遗传修饰(epigenetic modification)与表观遗传机制(epigenetic mechanism)等;(iv)非编码RNA与疾病关联,如人类疾病(human disease)、癌症病程(cancer progression)和癌症疗法(cancer therapy)等;(v)非编码RNA资源,如小RNA(small rnas)、长非编码RNA(long non-coding rnas)和小干扰RNA(small interference rnas)等;(vi)非编码RNA相关技术及其应用,如RNA干扰(rna interference)、治疗靶标(therapeutic targets)和潜在生物标志物(potential biomarkers)等。
通过考察词频的时间分布,将其中的突发词从大量的主题词中探测出来,依靠词频的变动趋势,能够确定学科研究的前沿领域和发展趋势。从近15年的突发词所组成的时间轴(图5)可以观察到每一年ncRNA的研究前沿变化情况。2000–2004年,该领域的研究方向仍聚焦于16SRNA、rRNA和tRNA等“传统”非编码RNA;从2005年开始,RNA干扰技术获得了人们更多的关注与青睐;2008年,科研人员又将目光转向了小RNA分子、miRNA的生物学机制、miRNA与基因的关系以及ncRNA对免疫系统的影响;2010年,聚焦在非编码RNA在乳腺癌中的生物学机制及其对神经系统的影响;2011年,miRNA的表达与调控研究和ncRNA在多能干细胞中的作用研究趋热;2013年之后,疾病的早期诊断、长非编码RNA、免疫应答和治疗靶点演化为为新一轮的研究趋势。
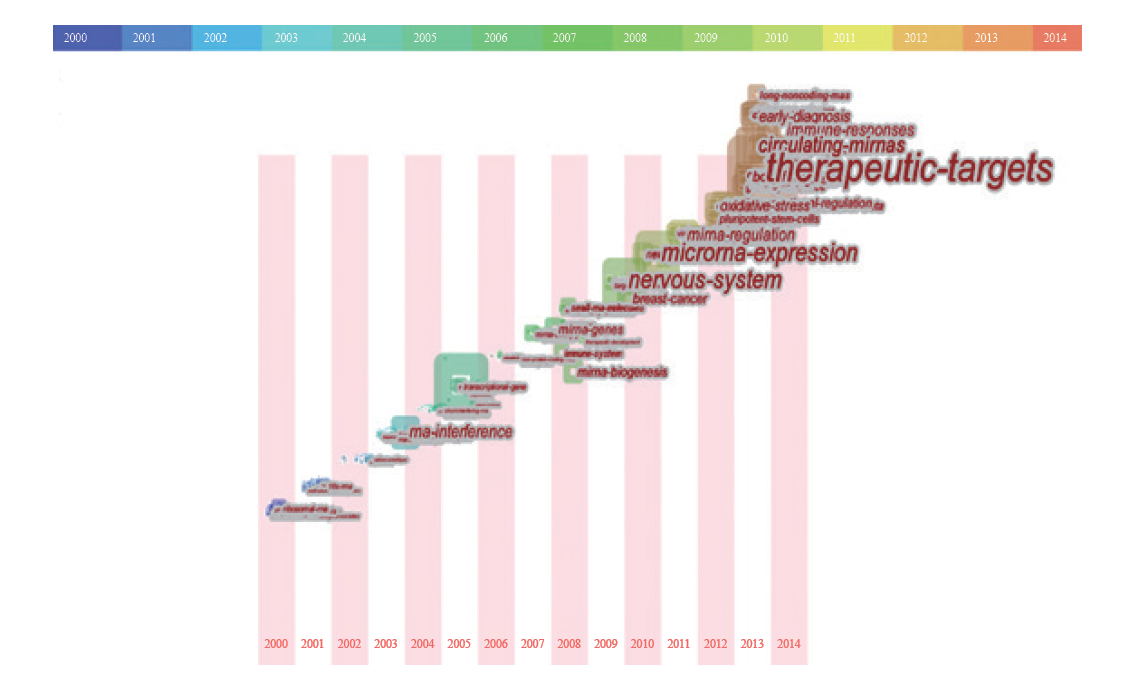
综上所述,近年来非编码RNA领域的前沿变化特点包括:(i)研究对象丰富多样,从rRNA、tRNA等“传统”非编码RNA发展到以miRNA、lncRNA和circRNA等种类与功能多元化的“现代”非编码RNA;(ii)研究角度层次分明,既开展非编码RNA的系统识别、功能确定等基础研究,又进行非编码RNA资源平台建设及相关技术的开发;(iii)研究目标向应用转移,从非编码RNA的分子机制到以临床治疗、药物开发为导向的转化医学研究,该学科的研究目标与人类的健康需求结合得越发密切。
3.5 前沿热点进展
非编码RNA研究领域的重大突破与热点进展集中在miRNA与lncRNA这“一短一长”两个研究子领域,下面就它们近年来的研究进展展开综述。
(1)miRNA近年研究进展
miRNA在动物的生长、发育中起着重要作用。2011年,Kim等在研究中发现了基于一种miRNA预测人类多能干细胞(hPSCs)向神经细胞分化命运的方法,证明miR-371-3有可能在人类多能干细胞神经性分化行为过程中发挥了关键性的作用,这为预测及控制多能干细胞神经分化命运提供了一个有潜力的作用因子[40]。随后,Colas等发现microRNA是胚胎发育时细胞命运的有力调控者[41]。Boon等发现miR-34a的表达与衰老诱导有关,并通过调控心脏的衰老过程,将miRNA、衰老和心脏功能联系在了一起[42]。以美国辛辛那提儿童医院为首的诸多机构通过研究发现,抑制LET-7这种小RNA发挥作用,可以让大脑神经元维持在“年轻”的状态[43,44]。Chen等破译了小鼠体内的一个“切换开关”——miR-155,发现其可以显著促进脂肪燃烧[45]。Pedersen等揭示了线虫中miR-79 调控神经发育的机制,研究显示其功能失常会造成线虫神经系统缺陷[46]。Tan等证实miR-128是神经细胞兴奋性和运动活性的最强调节物之一,而且是通过调节一种神经元信号途径来发挥作用的[47]。Zhang等通过实验证实在肌肉分化过程中,miR-1直接增强了线粒体基因的表达[48]。
miRNA不仅能够调控动物的生长发育,对于植物的开花、抗逆、增产等方面也有着重要的影响。曹晓风等通过对负责加工产生小分子RNA的酶OsDCL3a的研究,揭示依赖OsDCL3a的24-nt小分子RNA主要通过调控转座子旁临基因的表达进而对水稻重要农艺性状精细调控[49]。王佳伟等揭示了miRNA调控多年生草本植物弯曲碎米芥(Cardamine flexuosa)成花诱导的分子机理[50]。Zhang等发现,microRNA OsmiR397过表达能够增加谷粒的大小,促进圆锥花序分枝,从而提高水稻的产量[51]。Robert 等发现植物要生成生殖细胞,必须首先删除附着在全基因组 DNA 上的一系列表观遗传标记,导致新形成的生殖细胞置于遭受遗传损害的巨大危险之下,而miRNA通过将转座子维持在失活状态以规避这种风险[52]。
除此之外,人们更为关注的是miRNA与疾病/发病机制之间的紧密联系。Li等报道了miR-196b在混合性白血病中的新作用,指出了miRNA在肿瘤发生过程中的重要功能及其复杂的调控机制[53]。Wang等发现了miR-122在乙肝病毒复制以及持续感染途经中的新调控作用,为进一步了解HBV病毒的持续感染机制和肝癌发生途径提供了新的依据和阐释[54]。Dvinge等发文称人体自身免疫系统对抗乳腺癌是由小分子RNA控制,而后者对不同乳腺癌亚型的影响是不一样的[55]。Hasuwa等发现没有特殊类型小RNA的小鼠不会排卵,这一发现可能有一天会指导对人类不孕症的治疗[56]。Kornfeld等发现miR-802与胰岛素耐受性相关,该发现或为开发2型糖尿病新疗法提供思路[57]。Delorme-Axford发现胎盘microRNA能够保护胎儿不受病毒感染[58]。Xu等证实存在一种从前未知的TXNIP/miR-204/MAFA信号通路,其下调了胰岛素生成,推动了糖尿病的发生[59]。Peng等发现,肺癌中的microRNA-486是一种强效的抑癌分子,有助于调节肺癌细胞的增殖和迁移,并且诱导这些癌细胞程序性细胞死亡或细胞凋亡[60]。Chivukula等发现一个被认为在抑制结肠癌中起重要作用的 miRNA 簇——miR-143/145对肠伤口愈合至关重要[61]。
基于miRNA的重要性,研究人员对其检测方法、疾病治疗等应用型研究也颇为关注。Sundaram等鉴定出miR-198能够控制皮肤细胞转移,这是伤口愈合所必需的,因此其将成为开发减少或预防慢性伤口的新方法的关键[62]。Langlois提出了一种miRNA标靶技术,为流感病毒研究加设安全措施,以降低人类因接触实验室病毒而受感染的风险[63]。Hindson等证明液滴数字PCR(Droplet Digital PCR,ddPCR)技术在不同情况下可被用于准确、可重复性的血浆和血清 miRNA量化检测,从而为 miRNA 及其他核酸用于循环生物标志物的应用铺垫了道路[64]。Schirle等描绘出了miRNA在原子水平的运作,大大增进了人们对于生物学中基本调控系统的认识,并加速开发出一些利用其能力的新疗法[65]。Schultz等已经在全血中发现了诊断性的微 RNA 检测指标组,通过它们能够在一定程度上对病患是否患有胰腺癌进行甄别[66,67]。Slack等发现了一种新型的传送平台,利用肿瘤微环境的独特特性,让类似于miRNA镜像的反义分子进入到癌细胞中,形成了一种以miRNA为基础的抗癌药物以及靶向性药物传递的开发新模式[68]。
(2)lncRNA近年研究进展
lncRNA在动植物的生长、发育及分子调控过程中均起着非常重要的作用。lncRNA通过表达作用、结合蛋白质因子及调节染色质结构,可以参与增强子功能。Orom等特别对此方面的进展做出综述[69]。Lai也详细描述了长链非编码RNA激活子促进基因表达的机制,这些非编码RNA激活子(non-coding RNA-activators,ncRNA-a)在胚胎发育早期过程中对某些基因开关起至关重要的作用[70]。Klattenhoff等发现一种新型lncRNA——Braveheart(Bvht)——在哺乳动物发育过程与心血管发育谱系维持方面扮演了重要角色[71]。Sun等发现了一种反义长链非编码核糖核酸COOLAIR能够影响植物的开花时间[72]。Wang等发现了lincRNA-RoR在胚胎干细胞和iPS自我更新调控环路中起到关键作用,并由此指出了一种调控胚胎干细胞维持和分化的反馈环路[73,74]。Xue等发现生物钟同样受到lncRNA分子的调控[75]。曹雪涛等发现一种名为lnc-DC的lncRNA能够控制人类树突状细胞的分化[76]。
与miRNA类似,lncRNA和诸多人类疾病密切相关,并可以作为相应的药物靶点。Ramos等发现一种lncRNA在大脑发育中发挥了重要的作用,并有可能与几种毁灭性的神经系统疾病相关[77]。Yang等揭示了两种 lncRNAs可以阻止由于雄激素受体突变导致抵抗激素疗法的前列腺癌细胞的生长[78]。Nakagawa等发现一个lncRNA在某些情况下可能对于生育起着至关重要的作用[79]。Li等发现,在病原体入侵机体时,处在“激活”状态的巨噬细胞会生成一种含有lincRNA的新型复合体。这一复合体参与了免疫应答的调控,并且与川崎病(Kawasaki disease)有关[80]。Trimarchi等利用先进的遗传扫描技术鉴别出了6 023种长链非编码RNA,采用化学方法阻断其中LUNAR1的作用,可以遏制白血病的进展,该lncRNA可能成为治疗这种疾病的新型药物靶点[81]。Han等发现了一个此前未知的心脏lncRNA分子,其有可能是治疗和预防心力衰竭的关键[82]。Yang等揭示了lncRNA 通过调控肿瘤细胞瓦伯格效应(Warburg effect)促进肿瘤生长的作用机制[83]。
4 建议
随着各国不断推出非编码RNA相关的重大计划与资金资助,该领域逐渐成为生命科学研究中的热点,正在形成一门从基因组水平研究细胞中非编码RNA结构与功能的学科——“RNA组学”。通过对美国、欧盟、日本和中国相关政策规划的了解和学术论文的统计,本文对该领域的发展提出以下建议供参考。
4.1 加强战略部署,纳入重点计划
通过背景政策的了解,本文发现美国和日本从国家层面对该领域的发展进行了部署,采用的方式是加大力度支持本国牵头的专项国际合作计划(如ENCODE计划和FANTOM计划),开展持续性的研究性计划与资助辅助学科领域的进一步拓宽与深入。
在当前科技体制改革的大背景下,我国应当早日将非编码RNA研究纳入“国家重点研发计划”。同时,明确学科的研究目标与方向,从总体导向上对其做出规划。此外,要加强国际性的大型合作,推动计划做大做强。对学科的重视程度,不仅要从项目资助的角度去调整,还要从配套基础设施的配置、区域性研究网络的构建及研究型人才的培养等方面综合考量。
4.2 发展领域优势,补强技术短板
我国在非编码RNA领域已经形成了一些具有国际竞争力的优势方向,要不断加强此方面的研究。与此同时,在相关设备研究与技术开发方面从国外吸取经验,既可以通过这种基础与研发的“两栖”发展模式补强我国非编码RNA技术的短板,防止国外通过知识产权等形式对我国形成技术壁垒,又可以促进我国的技术研究,提升该领域的科技综合实力。
4.3 立足基础研究,开展转化医学研究
在非编码RNA基础研究的基础上,结合临床诊疗的需求,开展转化医学研究。将非编码RNA在人类疾病中的关键角色转化为重大疾病、神经退行性疾病和慢性病的治疗策略,开发以非编码RNA为基础的治疗药物、高效疗法等新型治疗模式,最终达到揭示非编码RNA在疾病中的作用目的的同时,发现相关疾病生物标记物,并展开药物研发和临床治疗。
4.4 建设数据平台,推动开放共享
随着生物大数据时代的到来,在非编码RNA领域应加强符合我国国情、具有我国特色的RNA数据中心与数据资源平台的建设,促进数据共享,加快数据转化为可改善健康的知识、产品和手段的步伐,同时保护研究参与者的隐私。数据平台可以通过相关数据的汇交、存储、加工和服务,实现多维数据的资源整合与开放共享,从而为科研部门、医疗机构等众多单位提供数据支撑和服务,让研究人员和患者共同受益。
致谢
中国科学院上海生命科学研究院生物化学与细胞生物学研究所王恩多院士、中山大学屈良鹄教授等专家对本文进行了审阅并提出了宝贵的修改意见和建议,谨致谢忱!
The authors have declared that no competing interests exist.
References
| [1] |
|
| [2] |
|
| [3] |
DOI:10.1038/35888
URL
[Cite within: 2]
|
| [4] |
|
| [5] |
URL
[Cite within: 1]
历史表明,每当DNA研究取得重大突破后,都会出现一个RNA研究的高潮.1953年DNA双螺旋结构的解析掀起了在RNA转录和翻译水平解读遗传信息的高潮,导致mRNA,tRNA和rRNA的发现以及遗传密码和“中心法则”的建立.1977年分裂基因(split gene)的发现极大地促进了在RNA转录后加工水平解读遗传信息表达的过程及机制.
|
| [6] |
DOI:10.1016/0092-8674(78)90248-9
PMID:688386
URL
[Cite within: 1]
Abstract The interruptions in the chicken ovalbumin gene which were reported previously (Breathnach, Mandel and Chambon, 1977) are shown to be due to the presence of intervening sequences which separate the messenger-coding sequences. We present evidence for an additional interruption of the gene, which, together with those reported earlier and by Garapin et al. (1978b), make a total of six intervening sequences. All of these intervening sequences are located in the DNA region that corresponds to the part of the ov mRNA which codes for amino acids. The seven coding fragments of the split ovalbumin gene are arranged in the same order and relative orientation as in the ovalbumin double-stranded cDNA. All the sequences coding for ov mRNA are contained in a chromosomal DNA region of 6000 bp, which is more than 3 times longer than ov mRNA. The general organization of the ovalbumin split gene is discussed.
|
| [7] |
DOI:10.1016/0092-8674(86)90064-4
PMID:3757028
URL
[Cite within: 1]
Indirect evidence suggests that the 5′ end of U1 snRNA recognizes the 5′ splice site in mRNA precursors by complementary base pairing. To test this hypothesis, we asked whether point mutations in the alternative 12S and 13S 5′ splice sites of the adenovirus E1A gene can be suppressed by compensatory base changes in human U1 snRNA. When the mutant E1A and U1 genes are cotransfected into HeLa cells, we observe efficient suppression of one mutation at position +5 in the 12S splice site, but exceedingly weak suppression of another mutation at position +3 in the 13S splice site. These and other results suggest that base pairing between U1 and the 5′ splice site is necessary but not sufficient for the splicing of mRNA precursors.
|
| [8] |
DOI:10.1016/0092-8674(87)90564-2
PMID:3552247
URL
[Cite within: 1]
The U2 snRNP binds to the site of branch formation during splicing of mammalian pre-mRNA in vitro. In Saccharomyces cerevisiae the branch site is within the so-called TACTAAC box (UACUAAC box), an absolutely conserved intron sequence required for splicing. Based on the identification and sequence of a U2 analogue in yeast, a specific base pairing interaction between the UACUAAC box and a highly conserved region of this snRNA can be proposed. To test this hypothesis, we have taken advantage of two mutations constructed previously in the UACUAAC box of an actin- HIS4 fusion. These mutant strains were transformed with stable plasmids bearing U2-like snRNAs into which changes predicted to restore base pairing had been introduced. Aliele-specific suppression of biological and biochemical phenotypes was observed in both cases. Recognition of the UACUAAC box thus relies, at least in part, on Watson-Crick base pairing with the yeast U2 analogue.
|
| [9] |
DOI:10.1016/0092-8674(82)90414-7
PMID:6297745
URL
[Cite within: 1]
In the macronuclear rRNA genes of Tetrahymena thermophila, a 413 bp intervening sequence (IVS) interrupts the 26S rRNA-coding region. A restriction fragment of the rDNA containing the IVS and portions of the adjacent rRNA sequences (exons) was inserted downstream from the lac UV5 promoter in a recombinant plasmid. Transcription of this template by purified Escherichia coli RNA polymerase in vitro produced a shortened version of the pre-rRNA, which was then deproteinized. When incubated with monovalent and divalent cations and a guanosine factor, this RNA underwent splicing. The reactions that were characterized included the precise excision of the IVS, attachment of guanosine to the 5' end of the IVS, covalent cyclization of the IVS and ligation of the exons. We conclude that splicing activity is intrinsic to the structure of the RNA, and that enzymes, small nuclear RNAs and folding of the pre-rRNA into an RNP are unnecessary for these reactions. We propose that the IVS portion of the RNA has several enzyme-like properties that enable it to break and reform phosphodiester bonds. The finding of autocatalytic rearrangements of RNA molecules has implications for the mechanism and the evolution of other reactions that involve RNA.
|
| [10] |
DOI:10.1016/0092-8674(86)90063-2
PMID:3019552
URL
[Cite within: 1]
The mitochondrial cytochrome oxidase (cox) subunit II gene from trypanosomes contains a frameshift at amino acid 170. This gene is highly conserved in different trypanosome species, suggesting that it is functional. Sequence determination of coxll transcripts of T. brucei and C. fasciculata reveals four extra, reading frame-restoring nucleotides at the frameshift position that are not encoded in the DNA. Southern blot analysis of DNA of both trypanosome species failed to show the existence of a second version of the coxll gene. We conclude, therefore, that the extra nucleotides are added during or after transcription of the frameshift gene by an RNA-editing process.
|
| [11] |
|
| [12] |
|
| [13] |
DOI:10.1016/0092-8674(93)90529-Y
PMID:8252621
URL
[Cite within: 1]
lin-4 is essential for the normal temporal control of diverse postembryonic developmental events in C. elegans. lin-4 acts by negatively regulating the level of LIN-14 protein, creating a temporal decrease in LIN-14 protein starting in the first larval stage (L1). We have cloned the C. elegans lin-4 locus by chromosomal walking and transformation rescue. We used the C. elegans clone to isolate the gene from three other Caenorhabditis species; all four Caenorhabditis clones functionally rescue the lin-4 null allele of C. elegans. Comparison of the lin-4 genomic sequence from these four species and site-directed mutagenesis of potential open reading frames indicated that lin-4 does not encode a protein. Two small lin-4 transcripts of approximately 22 and 61 nt were identified in C. elegans and found to contain sequences complementary to a repeated sequence element in the 3' untranslated region (UTR) of lin-14 mRNA, suggesting that lin-4 regulates lin-14 translation via an antisense RNA-RNA interaction.
|
| [14] |
DOI:10.1038/35002607
URL
[Cite within: 1]
|
| [15] |
DOI:10.1126/science.1064921
PMID:11679670
URL
[Cite within: 1]
In Caenorhabditis elegans, lin-4 and let-7 encode 22- and 21-nucleotide (nt) RNAs, respectively, which function as key regulators of developmental timing. Because the appearance of these short RNAs is regulated during development, they are also referred to as small temporal RNAs (stRNAs). We show that many 21- and 22-nt expressed RNAs, termed microRNAs, exist in invertebrates and vertebrates and that some of these novel RNAs, similar to let-7 stRNA, are highly conserved. This suggests that sequence-specific, posttranscriptional regulatory mechanisms mediated by small RNAs are more general than
|
| [16] |
|
| [17] |
|
| [18] |
DOI:10.2307/3869076
URL
[Cite within: 1]
We attempted to overexpress chalcone synthase (CHS) in pigmented petunia petals by introducing a chimeric petunia CHS gene. Unexpectedly, the introduced gene created a block in anthocyanin biosynthesis. Forty-two percent of plants with the introduced CHS gene produced totally white flowers and/or patterned flowers with white or pale nonclonal sectors on a wild-type pigmented background; none of hundreds of transgenic control plants exhibited such phenotypes. Progeny testing of one plant demonstrated that the novel color phenotype co-segregated with the introduced CHS gene; progeny without this gene were phenotypically wild type. The somatic and germinal stability of the novel color patterns was variable. RNase protection analysis of petal RNAs isolated from white flowers showed that, although the developmental timing of mRNA expression of the endogenous CHS gene was not altered, the level of the mRNA produced by this gene was reduced 50-fold from wild-type levels. Somatic reversion of plants with white flowers to phenotypically parental violet flowers was associated with a coordinate rise in the steady-state levels of the mRNAs produced by both the endogenous and the introduced CHS genes. Thus, in the altered white flowers, the expression of both genes was coordinately suppressed, indicating that expression of the introduced CHS gene was not alone sufficient for suppression of endogenous CHS transcript levels. The mechanism responsible for the reversible co-suppression of homologous genes in trans is unclear, but the erratic and reversible nature of this phenomenon suggests the possible involvement of methylation.
|
| [19] |
DOI:10.1111/j.1365-2958.1992.tb02202.x
PMID:1484489
URL
[Cite within: 1]
Summary Up to 36% of Neurospora crassa transformants showing an albino phenotype were recovered by transforming a wild-type strain with different portions of the carotenogenic albino-3 (al-3) and albino-1 (al-1) genes. The presence of the exogenous sequences (which were randomly integrated in ectopic locations) provoked a severe impairment in the expression of the endogenous al-1 or al-3 genes. This phenomenon, which we have termed‘quelling’, was found to be spontaneously and progressively reversible, leading to wild-type or intermediate phenotypes. The phenotypic reversion is characterized by a progressive release of the transcriptional inhibition and seems to correlate with a reduction of the number of the ectopic integrated sequences. Moreover, quelling appears to be monodirectional, as, once relieved, it cannot take place again, despite the continuing presence of some of the ectopic sequences in the genome.
|
| [20] |
DOI:10.1016/0092-8674(95)90082-9
PMID:7758115
URL
[Cite within: 1]
The first cleavage of C. elegans is asymmetric, generating daughter cells with different sizes, cytoplasmic components, and fates. Mutations in the par-1 gene disrupt this asymmetry. We report here that par-1 encodes a putative Ser/Thr kinase with similarity to kinases from yeasts and mammals. Two strong alleles have mutations in the kinase domain, suggesting that kinase activity is essential for par-1 function. PAR-1 protein is localized to the posterior periphery of the zygote and is distributed in a polar fashion preceding the asymmetric divisions of the germline lineage. Because PAR-1 distribution in the germline correlates with the distribution of germline-specific P granules, it is possible that PAR-1 functions in germline development as well as in establishing embryonic polarity.
|
| [21] |
DOI:10.1038/nature04916
PMID:16751777
URL
[Cite within: 1]
Abstract Small RNAs bound to Argonaute proteins recognize partially or fully complementary nucleic acid targets in diverse gene-silencing processes. A subgroup of the Argonaute proteins--known as the 'Piwi family'--is required for germ- and stem-cell development in invertebrates, and two Piwi members--MILI and MIWI--are essential for spermatogenesis in mouse. Here we describe a new class of small RNAs that bind to MILI in mouse male germ cells, where they accumulate at the onset of meiosis. The sequences of the over 1,000 identified unique molecules share a strong preference for a 5' uridine, but otherwise cannot be readily classified into sequence families. Genomic mapping of these small RNAs reveals a limited number of clusters, suggesting that these RNAs are processed from long primary transcripts. The small RNAs are 26-31 nucleotides (nt) in length--clearly distinct from the 21-23 nt of microRNAs (miRNAs) or short interfering RNAs (siRNAs)--and we refer to them as 'Piwi-interacting RNAs' or piRNAs. Orthologous human chromosomal regions also give rise to small RNAs with the characteristics of piRNAs, but the cloned sequences are distinct. The identification of this new class of small RNAs provides an important starting point to determine the molecular function of Piwi proteins in mammalian spermatogenesis.
|
| [22] |
DOI:10.1038/nature04917
PMID:16751776
URL
[Cite within: 1]
Abstract Small RNAs associate with Argonaute proteins and serve as sequence-specific guides to regulate messenger RNA stability, protein synthesis, chromatin organization and genome structure. In animals, Argonaute proteins segregate into two subfamilies. The Argonaute subfamily acts in RNA interference and in microRNA-mediated gene regulation using 21-22-nucleotide RNAs as guides. The Piwi subfamily is involved in germline-specific events such as germline stem cell maintenance and meiosis. However, neither the biochemical function of Piwi proteins nor the nature of their small RNA guides is known. Here we show that MIWI, a murine Piwi protein, binds a previously uncharacterized class of approximately 29-30-nucleotide RNAs that are highly abundant in testes. We have therefore named these Piwi-interacting RNAs (piRNAs). piRNAs show distinctive localization patterns in the genome, being predominantly grouped into 20-90-kilobase clusters, wherein long stretches of small RNAs are derived from only one strand. Similar piRNAs are also found in human and rat, with major clusters occurring in syntenic locations. Although their function must still be resolved, the abundance of piRNAs in germline cells and the male sterility of Miwi mutants suggest a role in gametogenesis.
|
| [23] |
DOI:10.1016/j.tig.2005.03.007
PMID:15851066
URL
[Cite within: 1]
Abstract The past four years have seen an explosion in the number of detected RNA transcripts with no apparent protein-coding potential. This has led to speculation that non-protein-coding RNAs (ncRNAs) might be as important as proteins in the regulation of vital cellular functions. However, there has been significantly less progress in actually demonstrating the functions of these transcripts. In this article, we review the results of recent experiments that show that transcription of non-protein-coding RNA is far more widespread than was previously anticipated. Although some ncRNAs act as molecular switches that regulate gene expression, the function of many ncRNAs is unknown. New experimental and computational approaches are emerging that will help determine whether these newly identified transcription products are evidence of important new biochemical pathways or are merely 'junk' RNA generated by the cell as a by-product of its functional activities.
|
| [24] |
DOI:10.1016/0166-6851(90)90215-8
PMID:360709
URL
[Cite within: 1]
Abstract The mouse H19 gene was identified as an abundant hepatic fetal-specific mRNA under the transcriptional control of a trans-acting locus termed raf. The protein this gene encoded was not apparent from an analysis of its nucleotide sequence, since the mRNA contained multiple translation termination signals in all three reading frames. As a means of assessing which of the 35 small open reading frames might be important to the function of the gene, the human H19 gene was cloned and sequenced. Comparison of the two homologs revealed no conserved open reading frame. Cellular fractionation showed that H19 RNA is cytoplasmic but not associated with the translational machinery. Instead, it is located in a particle with a sedimentation coefficient of approximately 28S. Despite the fact that it is transcribed by RNA polymerase II and is spliced and polyadenylated, we suggest that the H19 RNA is not a classical mRNA. Instead, the product of this unusual gene may be an RNA molecule.
|
| [25] |
DOI:10.1016/0092-8674(92)90519-I
PMID:1423610
URL
[Cite within: 1]
The Xist gene maps to the X inactivation center region in both mouse and human, and previous analysis of the 3' end of the gene has demonstrated inactive X-specific expression, suggesting a possible role in X inactivation. We have now analyzed the entire mouse Xist gene. The mature inactive X-specific transcript is 15 kb in length and contains no conserved ORF. The Xist sequence contains a number of regions comprised of tandem repeats. Comparison with the human XIST gene demonstrates significant conservation of sequence and gene structure. Xist RNA is not associated with the translational machinery of the cell and is located almost exclusively in the nucleus. Together with conservation of inactive X-specific expression, these findings support a role for Xist in X inactivation, possibly as a functional RNA or as a chromatin organizer region.
|
| [26] |
URL
[Cite within: 1]
非编码RNA(noncoding RNA,ncRNA)是指不被翻译成蛋白质的一类RNA,近几年来关于它们的功能研究越来越引起人们的重视.现在已经发现了一些中小型ncRNA,比如microRNA、snoRNA、tRNA等,但是关于长ncRNA(lncRNA)的研究还不够完善.本篇综述回顾了ncRNA特别是lncRNA的生物信息学研究进展,包括它们的研究历程、基本特点、与疾病的关系,以及对已有的预测非编码RNA的计算机方法进行了分析和比较,并且介绍了利用机器学习模型整合新一代高通量测序数据的方法.
|
| [27] |
|
| [28] |
DOI:10.1016/j.cell.2007.05.022
PMID:2084369
URL
[Cite within: 1]
Noncoding RNAs (ncRNA) participate in epigenetic regulation but are poorly understood. Here we characterize the transcriptional landscape of the four human HOX loci at five base pair resolution in 11 anatomic sites and identify 231 HOX ncRNAs that extend known transcribed regions by more than 30 kilobases. HOX ncRNAs are spatially expressed along developmental axes and possess unique sequence motifs, and their expression demarcates broad chromosomal domains of differential histone methylation and RNA polymerase accessibility. We identified a 2.2 kilobase ncRNA residing in the HOXC locus, termed HOTAIR, which represses transcription in trans across 40 kilobases of the HOXD locus. HOTAIR interacts with Polycomb Repressive Complex 2 (PRC2) and is required for PRC2 occupancy and histone H3 lysine-27 trimethylation of HOXD locus. Thus, transcription of ncRNA may demarcate chromosomal domains of gene silencing at a distance; these results have broad implications for gene regulation in development and disease states.
|
| [29] |
DOI:10.1038/7734
URL
[Cite within: 1]
|
| [30] |
DOI:10.1126/science.1197349
PMID:21127216
URL
[Cite within: 1]
Abstract Vernalization is an environmentally-induced epigenetic switch in which winter cold triggers epigenetic silencing of floral repressors and thus provides competence to flower in spring. In Arabidopsis, winter cold triggers enrichment of tri-methylated histone H3 Lys(27) at chromatin of the floral repressor, FLOWERING LOCUS C (FLC), and results in epigenetically stable repression of FLC. This epigenetic change is mediated by an evolutionarily conserved repressive complex, polycomb repressive complex 2 (PRC2). Here, we show that a long intronic noncoding RNA [termed COLD ASSISTED INTRONIC NONCODING RNA (COLDAIR)] is required for the vernalization-mediated epigenetic repression of FLC. COLDAIR physically associates with a component of PRC2 and targets PRC2 to FLC. Our results show that COLDAIR is required for establishing stable repressive chromatin at FLC through its interaction with PRC2.
|
| [31] |
DOI:10.1016/j.molcel.2010.03.019
PMID:20542000
URL
[Cite within: 1]
Abstract Polycomb proteins maintain cell identity by repressing the expression of developmental regulators specific for other cell types. Polycomb repressive complex-2 (PRC2) catalyzes trimethylation of histone H3 lysine-27 (H3K27me3). Although repressed, PRC2 targets are generally associated with the transcriptional initiation marker H3K4me3, but the significance of this remains unclear. Here, we identify a class of short RNAs, approximately 50-200 nucleotides in length, transcribed from the 5' end of polycomb target genes in primary T cells and embryonic stem cells. Short RNA transcription is associated with RNA polymerase II and H3K4me3, occurs in the absence of mRNA transcription, and is independent of polycomb activity. Short RNAs form stem-loop structures resembling PRC2 binding sites in Xist, interact with PRC2 through SUZ12, cause gene repression in cis, and are depleted from polycomb target genes activated during cell differentiation. We propose that short RNAs play a role in the association of PRC2 with its target genes. Copyright (c) 2010 Elsevier Inc. All rights reserved.
|
| [32] |
|
| [33] |
|
| [34] |
DOI:10.1016/0092-8674(76)90223-3
PMID:182384
URL
[Cite within: 1]
When passaged at high multiplicity, four strains of Sendai virus all showed evidence that they contained defective interfering (DI) particles. RNA isolated from nucleocapsids of cells infected with the high multiplicity passage stocks was found to consist of only minor amounts of nondefective genome length RNA and major amounts of smaller RNAs, the DI-RNAs. These DI-RNAs were found to have unusual and variable sedimentation properties in sucrose gradients, but were found to represent unique segments of the viral genome by length measurements in the electron microscope and by hybridization. A striking feature of the DI-RNAs is their ability to form circular structures, indicating that the ends of the DI-RNA are complementary. The implications of this finding in terms of the mechanism of genome replication is discussed.
|
| [35] |
DOI:10.1073/pnas.73.11.3852
PMID:1069269
URL
[Cite within: 1]
Abstract Viroids are uncoated infectious RNA molecules pathogenic to certain higher plants. Four different highly purified viroids were studied. By ultracentrifugation, thermal denaturation, electron microscopy, and end group analysis the following features were established: (i) the molecular weight of cucumber pale fruit viroid from tomato is 110,000, of citrus exocortis viroid from Gynura 119,000, of citrus exocortis viroid from tomato 119,000 and of potato spindle tuber viroid from tomato 127,000. (ii) Viroids are single-stranded molecules. (iii) Virods exhibit high thermal stability, cooperativity, and self-complementarity resulting in a rod-like native structure. (iv) Viroids are covalently closed circular RNA molecules.
|
| [36] |
DOI:10.1073/pnas.87.19.7628
PMID:1699230
URL
[Cite within: 1]
Circular RNA replicons have been reported in plants and, in one case, in animal cells. We describe such an element in yeast. In certain yeast strains, a 20S RNA species appears on transfer of cells to acetate medium. This phenotype shows cytoplasmic (non-Mendelian) inheritance and the 20S RNA is associated with 23-kDa protein subunits as a 32S particle. We demonstrate that yeast 20S RNA is an independent replicon with no homology to host genomic, mitochondrial, or 2-microns plasmid DNA or to the L-A, L-BC, or M1 double-stranded RNA viruses of yeast. The circularity of the 20S RNA is shown by the apparent absence of 3' and 5' ends, by two-dimensional gel electrophoresis, and by electron microscopy. Replication of yeast 20S RNA proceeds through an RNA-RNA pathway, and a 10,000-fold amplification occurs on shift to acetate medium. The copy number of 20S RNA is also reduced severalfold by the SKI gene products, a host antiviral system that also lowers the copy numbers of yeast double-stranded RNA viruses. Yeast 20S RNA and the hepatitis delta virus show some similarities.
|
| [37] |
|
| [38] |
DOI:10.1093/nar/gkr1009
PMID:3326292962175459219598570825
URL
[Cite within: 1]
Circular RNA forms had been described in all domains of life. Such RNAs were shown to have diverse biological functions, including roles in the life cycle of viral and viroid genomes, and in maturation of permuted tRNA genes. Despite their potentially important biological roles, discovery of circular RNAs has so far been mostly serendipitous. We have developed circRNA-seq, a combined experimental/computational approach that enriches for circular RNAs and allows profiling their prevalence in a whole-genome, unbiased manner. Application of this approach to the archaeon Sulfolobus solfataricus P2 revealed multiple circular transcripts, a subset of which was further validated independently. The identified circular RNAs included expected forms, such as excised tRNA introns and rRNA processing intermediates, but were also enriched with non-coding RNAs, including C/D box RNAs and RNase P, as well as circular RNAs of unknown function. Many of the identified circles were conserved in Sulfolobus acidocaldarius, further supporting their functional significance. Our results suggest that circular RNAs, and particularly circular non-coding RNAs, are more prevalent in archaea than previously recognized, and might have yet unidentified biological roles. Our study establishes a specific and sensitive approach for identification of circular RNAs using RNA-seq, and can readily be applied to other organisms.
|
| [39] |
DOI:10.1371/journal.pone.0030733
PMID:3270023
URL
[Cite within: 1]
Most human pre-mRNAs are spliced into linear molecules that retain the exon order defined by the genomic sequence. By deep sequencing of RNA from a variety of normal and malignant human cells, we found RNA transcripts from many human genes in which the exons were arranged in a non-canonical order. Statistical estimates and biochemical assays provided strong evidence that a substantial fraction of the spliced transcripts from hundreds of genes are circular RNAs. Our results suggest that a non-canonical mode of RNA splicing, resulting in a circular RNA isoform, is a general feature of the gene expression program in human cells.
|
| [40] |
DOI:10.1016/j.stem.2011.04.002
PMID:21624813
URL
[Cite within: 1]
The use of pluripotent stem cells in regenerative medicine and disease modeling is complicated by the variation in differentiation properties between lines. In this study, we characterized 13 human embryonic stem cell (hESC) and 26 human induced pluripotent stem cell (hiPSC) lines to identify markers that predict neural differentiation behavior. At a general level, markers previously known to distinguish mouse ESCs from epiblast stem cells (EPI-SCs) correlated with neural differentiation behavior. More specifically, quantitative analysis of miR-371-3 expression prospectively identified hESC and hiPSC lines with differential neurogenic differentiation propensity and invivo dopamine neuron engraftment potential. Transient KLF4 transduction increased miR-371-3 expression and altered neurogenic behavior and pluripotency marker expression. Conversely, suppression of miR-371-3 expression in KLF4-transduced cells rescued neural differentiation propensity. miR-371-3 expression level therefore appears to have both a predictive and a functional role in determining human pluripotent stem cell neurogenic differentiation behavior.
|
| [41] |
DOI:10.1101/gad.200758.112
URL
[Cite within: 1]
Tight control over the segregation of endoderm, mesoderm, and ectoderm is essential for normal embryonic development of all species, yet how neighboring embryonic blastomeres can contribute to different germ layers has never been fully explained. We postulated that microRNAs, which fine-tune many biological processes, might modulate the response of embryonic blastomeres to growth factors and other signals that govern germ layer fate. A systematic screen of a whole-genome microRNA library revealed that the let-7 and miR-18 families increase mesoderm at the expense of endoderm in mouse embryonic stem cells. Both families are expressed in ectoderm and mesoderm, but not endoderm, as these tissues become distinct during mouse and frog embryogenesis. Blocking let-7 function in vivo dramatically affected cell fate, diverting presumptive mesoderm and ectoderm into endoderm. siRNA knockdown of computationally predicted targets followed by mutational analyses revealed that let-7 and miR-18 down-regulate Acvr1b and Smad2, respectively, to attenuate Nodal responsiveness and bias blastomeres to ectoderm and mesoderm fates. These findings suggest a crucial role for the let-7 and miR-18 families in germ layer specification and reveal a remarkable conservation of function from amphibians to mammals.
|
| [42] |
DOI:10.1038/nature11919
PMID:23426265
URL
[Cite within: 1]
Ageing is the predominant risk factor for cardiovascular diseases(1) and contributes to a significantly worse outcome in patients with acute myocardial infarction(2). MicroRNAs (miRNAs) have emerged as crucial regulators of cardiovascular function and some miRNAs have key roles in ageing(3,4). We propose that altered expression of miRNAs in the heart during ageing contributes to the age-dependent decline in cardiac function. Here we show that miR-34a is induced in the ageing heart and that in vivo silencing or genetic deletion of miR-34a reduces age-associated cardiomyocyte cell death. Moreover, miR-34a inhibition reduces cell death and fibrosis following acute myocardial infarction and improves recovery of myocardial function. Mechanistically, we identified PNUTS (also known as PPP1R10) as a novel direct miR-34a target, which reduces telomere shortening, DNA damage responses and cardiomyocyte apoptosis, and improves functional recovery after acute myocardial infarction. Together, these results identify age-induced expression of miR-34a and inhibition of its target PNUTS as a key mechanism that regulates cardiac contractile function during ageing and after acute myocardial infarction, by inducing DNA damage responses and telomere attrition.
|
| [43] |
DOI:10.1126/science.1231321
PMID:23599497
URL
[Cite within: 1]
Like mammalian neurons, Caenorhabditis elegans neurons lose axon regeneration ability as they age, but it is not known why. Here, we report that let-7 contributes to a developmental decline in anterior ventral microtubule (AVM) axon regeneration. In older AVM axons, let-7 inhibits regeneration by down-regulating LIN-41, an important AVM axon regeneration-promoting factor. Whereas let-7 inhibits lin-41 expression in older neurons through the lin-41 3' untranslated region, lin-41 inhibits let-7 expression in younger neurons through Argonaute ALG-1. This reciprocal inhibition ensures that axon regeneration is inhibited only in older neurons. These findings show that a let-7-lin-41 regulatory circuit, which was previously shown to control timing of events in mitotic stem cell lineages, is reutilized in postmitotic neurons to control postdifferentiation events.
|
| [44] |
DOI:10.1126/science.1237921
PMID:23599470
URL
[Cite within: 1]
A signaling pathway is implicated in the age-dependent decline of neuron regeneration. [Also see Report by Zou et al.]
|
| [45] |
DOI:10.1038/ncomms2742
PMID:23612310
URL
[Cite within: 1]
Brown adipocytes are a primary site of energy expenditure and reside not only in classical brown adipose tissue but can also be found in white adipose tissue. Here we show that microRNA 155 is enriched in brown adipose tissue and is highly expressed in proliferating brown preadipocytes but declines after induction of differentiation. Interestingly, microRNA 155 and its target, the adipogenic transcription factor CCAAT/enhancer-binding protein β, form a bistable feedback loop integrating hormonal signals that regulate proliferation or differentiation. Inhibition of microRNA 155 enhances brown adipocyte differentiation and induces a brown adipocyte-like phenotype (‘browning’) in white adipocytes. Consequently, microRNA 155-deficient mice exhibit increased brown adipose tissue function and ‘browning’ of white fat tissue. In contrast, transgenic overexpression of microRNA 155 in mice causes a reduction of brown adipose tissue mass and impairment of brown adipose tissue function. These data demonstrate that the bistable loop involving microRNA 155 and CCAAT/enhancer-binding protein β regulates brown lineage commitment, thereby, controlling the development of brown and beige fat cells.
|
| [46] |
DOI:10.1126/science.1242528
PMID:24052309
URL
[Cite within: 1]
Abstract An appropriate balance in glycosylation of proteoglycans is crucial for their ability to regulate animal development. Here, we report that the Caenorhabditis elegans microRNA mir-79, an ortholog of mammalian miR-9, controls sugar-chain homeostasis by targeting two proteins in the proteoglycan biosynthetic pathway: a chondroitin synthase (SQV-5; squashed vulva-5) and a uridine 5'-diphosphate-sugar transporter (SQV-7). Loss of mir-79 causes neurodevelopmental defects through SQV-5 and SQV-7 dysregulation in the epidermis. This results in a partial shutdown of heparan sulfate biosynthesis that impinges on a LON-2/glypican pathway and disrupts neuronal migration. Our results identify a regulatory axis controlled by a conserved microRNA that maintains proteoglycan homeostasis in cells.
|
| [47] |
DOI:10.1126/science.1244193
PMID:3932786
URL
[Cite within: 1]
The control of motor behavior in animals and humans requires constant adaptation of neuronal networks to signals of various types and strengths. We found that microRNA-128 (miR-128), which is expressed in adult neurons, regulates motor behavior by modulating neuronal signaling networks and excitability. miR-128 governs motor activity by suppressing the expression of various ion channels and signaling components of the extracellular signal-regulated kinase ERK2 network that regulate neuronal excitability. In mice, a reduction of miR-128 expression in postnatal neurons causes increased motor activity and fatal epilepsy. Overexpression of miR-128 attenuates neuronal responsiveness, suppresses motor activity, and alleviates motor abnormalities associated with Parkinson's-like disease and seizures in mice. These data suggest a therapeutic potential for miR-128 in the treatment of epilepsy and movement disorders.
|
| [48] |
DOI:10.1016/j.cell.2014.05.047
PMID:25083871
URL
[Cite within: 1]
Abstract MicroRNAs are well known to mediate translational repression and mRNA degradation in the cytoplasm. Various microRNAs have also been detected in membrane-compartmentalized organelles, but the functional significance has remained elusive. Here, we report that miR-1, a microRNA specifically induced during myogenesis, efficiently enters the mitochondria where it unexpectedly stimulates, rather than represses, the translation of specific mitochondrial genome-encoded transcripts. We show that this positive effect requires specific miR:mRNA base-pairing and Ago2, but not its functional partner GW182, which is excluded from the mitochondria. We provide evidence for the direct action of Ago2 in mitochondrial translation by crosslinking immunoprecipitation coupled with deep sequencing (CLIP-seq), functional rescue with mitochondria-targeted Ago2, and selective inhibition of the microRNA machinery in the cytoplasm. These findings unveil a positive function of microRNA in mitochondrial translation and suggest a highly coordinated myogenic program via miR-1-mediated translational stimulation in the mitochondria and repression in the cytoplasm. Copyright 2014 Elsevier Inc. All rights reserved.
|
| [49] |
DOI:10.1073/pnas.1318131111
URL
[Cite within: 1]
Transposable elements (TEs) and repetitive sequences make up over 35% of the rice (Oryza sativa) genome. The host regulates the activity of different TEs by different epigenetic mechanisms, including DNA methylation, histone H3K9 methylation, and histone H3K4 demethylation. TEs can also affect the expression of host genes. For example, miniature inverted repeat TEs (MITEs), dispersed high copy-number DNA TEs, can influence the expression of nearby genes. In plants, 24-nt small interfering RNAs (siRNAs) are mainly derived from repeats and TEs. However, the extent to which TEs, particularly MITEs associated with 24-nt siRNAs, affect gene expression remains elusive. Here, we show that the rice Dicer-like 3 homolog OsDCL3a is primarily responsible for 24-nt siRNA processing. Impairing OsDCL3a expression by RNA interference caused phenotypes affecting important agricultural traits; these phenotypes include dwarfism, larger flag leaf angle, and fewer secondary branches. We used small RNA deep sequencing to identify 535,054 24-nt siRNA clusters. Of these clusters, 82% were OsDCL3a-dependent and showed significant enrichment of MITEs. Reduction of OsDCL3a function reduced the 24-nt siRNAs predominantly from MITEs and elevated expression of nearby genes. OsDCL3a directly targets genes involved in gibberellin and brassinosteroid homeostasis; OsDCL3a deficiency may affect these genes, thus causing the phenotypes of dwarfism and enlarged flag leaf angle. Our work identifies OsDCL3a-dependent 24-nt siRNAs derived from MITEs as broadly functioning regulators for fine-tuning gene expression, which may reflect a conserved epigenetic mechanism in higher plants with genomes rich in dispersed repeats or TEs.
|
| [50] |
DOI:10.1126/science.1234340
PMID:23723237
URL
[Cite within: 1]
Plants flower in response to many varied cues, such as temperature, photoperiod, and age. The floral transition of Cardamine flexuosa, a herbaceous biennial-to-perennial plant, requires exposure to cold temperature, a treatment known as vernalization. C. flexuosa younger than 5 weeks old are not fully responsive to cold treatment. We demonstrate that the levels of two age-regulated microRNAs, miR156 and miR172, regulate the timing of sensitivity in response to vernalization. Age and vernalization pathways coordinately regulate flowering through modulating the expression of CfSOC1, a flower-promoting MADS-box gene. The related annual Arabidopsis thaliana, which has both vernalization and age pathways, does not possess an age-dependent vernalization response. Thus, the recruitment of age cue in response to environmental signals contributes to the evolution of life cycle in plants.
|
| [51] |
DOI:10.1038/nbt.2646
PMID:23873084
URL
[Cite within: 1]
Increasing grain yields is a major focus of crop breeders around the world. Here we report that overexpression of the rice microRNA (miRNA) OsmiR397, which is naturally highly expressed in young panicles and grains, enlarges grain size and promotes panicle branching, leading to an increase in overall grain yield of up to 25% in a field trial. To our knowledge, no previous report has shown a positive regulatory role of miRNA in the control of plant seed size and grain yield. We determined that OsmiR397 increases grain yield by downregulating its target, OsLAC, whose product is a laccase-like protein that we found to be involved in the sensitivity of plants to brassinosteroids. As miR397 is highly conserved across different species, our results suggest that manipulating miR397 may be useful for increasing grain yield not only in rice but also in other cereal crops.
|
| [52] |
DOI:10.1038/nature13069
PMID:4074602
URL
[Cite within: 1]
In plants, post-transcriptional gene silencing (PTGS) is mediated by DICER-LIKE1 (DCL1)-dependent miRNAs, that also trigger 21-nt secondary siRNA via RNA DEPENDENT RNA POLYMERASE6 (RDR6), DCL4, and ARGONAUTE1 (AGO1)1–3, while transcriptional gene silencing (TGS) of transposons is mediated by 24-nt heterochromatic (het)siRNA RDR2, DCL3 and AGO44. Transposons can also give rise to abundant 21-nt “epigenetically activated” small interfering RNAs (easiRNAs) in DECREASE IN DNA METHYLATION1 (ddm1) and DNA METHYLTRANSFERASE1 (met1) mutants, as well as in the vegetative nucleus of pollen grains5, and in dedifferentiated plant cell cultures6. Here we show that easiRNAs resemble secondary siRNAs, in that thousands of transposon transcripts are specifically targeted by more than fifty miRNAs for cleavage and processing by RDR6. Loss of RDR6, DCL4 or DCL1 in a ddm1 background results in loss of 21-nt easiRNA, and severe infertility, but 24-nt hetsiRNA are partially restored, supporting an antagonistic relationship between PTGS and TGS. Thus miRNA-directed easiRNA biogenesis is a latent mechanism that specifically targets transposon transcripts, but only when they are epigenetically reactivated during reprogramming of the germline. This ancient recognition mechanism may have been retained both by transposons to evade long-term heterochromatic silencing, and by their hosts for genome defence.
|
| [53] |
DOI:10.1038/ncomms1681
PMID:22353710
URL
[Cite within: 1]
HOXA9 and MEIS1 have essential oncogenic roles in mixed lineage leukaemia (MLL)-rearranged leukaemia. Here we show that they are direct targets of miRNA-196b, a microRNA (miRNA) located adjacent to and co-expressed with HOXA9, in MLL-rearranged leukaemic cells. Forced expression of miR-196b significantly delays MLL-fusion-mediated leukemogenesis in primary bone marrow transplantation through suppressing Hoxa9/Meis1 expression. However, ectopic expression of miR-196b results in more aggressive leukaemic phenotypes and causes much faster leukemogenesis in secondary transplantation than MLL fusion alone, likely through the further repression of Fas expression, a proapoptotic gene downregulated in MLL-rearranged leukaemia. Overexpression of FAS significantly inhibits leukemogenesis and reverses miR-196b-mediated phenotypes. Targeting Hoxa9/Meis1 and Fas by miR-196b is probably also important for normal haematopoiesis. Thus, our results uncover a previously unappreciated miRNA-regulation mechanism by which a single miRNA may target both oncogenes and tumour suppressors, simultaneously, or, sequentially, in tumourigenesis and normal development per cell differentiation, indicating that miRNA regulation is much more complex than previously thought.
|
| [54] |
DOI:10.1002/hep.24809
PMID:22105316
URL
[Cite within: 1]
Hepatitis B virus (HBV) causes chronic infection in about 350 million people worldwide. Given the important role of the most abundant liver-specific microRNA, miR-122, in hepatic function and liver pathology, here we investigated the potential role and mechanism of miR-122 in regulating HBV replication. We found that miR-122 expression in liver was significantly down-regulated in patients with HBV infection compared with healthy controls, and the miR-122 levels were negatively correlated with intrahepatic viral load and hepatic necroinflammation. The depletion of endogenous miR-122 by its antisense inhibitor led to enhanced HBV replication, whereas overexpression of miR-122 by transfection of mimic or its expression vector inhibited viral production. We next identified cyclin G(1) as an miR-122 target from multiple candidate target genes that are involved in the regulation of HBV replication. Overexpression and knockdown studies both showed that cyclin G(1) regulated viral replication in HBV transfected cells. We also observed that cyclin G(1) expression was up-regulated in HBV-infected patients, and cyclin G(1) levels were inversely associated with miR-122 expression in liver tissues. Using coimmunoprecipitation, a luciferase reporter system, and electrophoretic mobility shift assay, we further demonstrated that cyclin G(1) specifically interacted with p53, and this interaction blocked the specific binding of p53 to HBV enhancer elements and simultaneously abrogated p53-mediated inhibition of HBV transcription. Finally, we show that miR-122 suppressed HBV replication in p53 wildtype cells but not in null isogenic cells.miR-122 down-regulates its target cyclin G(1) , and thus interrupts the interaction between cyclin G(1) and p53 and abrogates p53-mediated inhibition of HBV replication. Our work shows that miR-122 down-regulation induced by HBV infection can impact HBV replication and possibly contribute to viral persistence and carcinogenesis.
|
| [55] |
DOI:10.1038/nature12108
PMID:23644459
URL
[Cite within: 1]
Abstract MicroRNAs (miRNAs) show differential expression across breast cancer subtypes, and have both oncogenic and tumour-suppressive roles. Here we report the miRNA expression profiles of 1,302 breast tumours with matching detailed clinical annotation, long-term follow-up and genomic and messenger RNA expression data. This provides a comprehensive overview of the quantity, distribution and variation of the miRNA population and provides information on the extent to which genomic, transcriptional and post-transcriptional events contribute to miRNA expression architecture, suggesting an important role for post-transcriptional regulation. The key clinical parameters and cellular pathways related to the miRNA landscape are characterized, revealing context-dependent interactions, for example with regards to cell adhesion and Wnt signalling. Notably, only prognostic miRNA signatures derived from breast tumours devoid of somatic copy-number aberrations (CNA-devoid) are consistently prognostic across several other subtypes and can be validated in external cohorts. We then use a data-driven approach to seek the effects of miRNAs associated with differential co-expression of mRNAs, and find that miRNAs act as modulators of mRNA-mRNA interactions rather than as on-off molecular switches. We demonstrate such an important modulatory role for miRNAs in the biology of CNA-devoid breast cancers, a common subtype in which the immune response is prominent. These findings represent a new framework for studying the biology of miRNAs in human breast cancer.
|
| [56] |
DOI:10.1126/science.1237999
PMID:23765281
URL
[Cite within: 1]
Ovulation in the mouse and other mammals is controlled by hormones secreted by the hypothalamo-pituitary-ovarian axis. We describe anovulation and infertility in female mice lacking the microRNAs miR-200b and miR-429. Both miRNAs are strongly expressed in the pituitary gland, where they suppress expression of the transcriptional repressor ZEB1. Eliminating these miRNAs, in turn, inhibits luteinizing hormone (LH) synthesis by repressing transcription of its -subunit gene, which leads to lowered serum LH concentration, an impaired LH surge, and failure to ovulate. Our results reveal roles for miR-200b and miR-429, and their target the Zeb1 gene, in the regulation of mammalian reproduction. Thus, the hypothalamo-pituitary-ovarian axis was shown to require miR-200b and miR-429 to support ovulation.
|
| [57] |
DOI:10.1038/nature11793
PMID:23389544
URL
[Cite within: 1]
Insulin resistance represents a hallmark during the development of type 2 diabetes mellitus and in the pathogenesis of obesity-associated disturbances of glucose and lipid metabolism(1-3). MicroRNA (miRNA)-dependent post-transcriptional gene silencing has been recognized recently to control gene expression in disease development and progression, including that of insulin-resistant type 2 diabetes. The deregulation of miRNAs miR-143 (ref. 4), miR-181 (ref. 5), and miR-103 and miR-107 (ref. 6) alters hepatic insulin sensitivity. Here we report that the expression of miR-802 is increased in the liver of two obese mouse models and obese human subjects. Inducible transgenic overexpression of miR-802 in mice causes impaired glucose tolerance and attenuates insulin sensitivity, whereas reduction of miR-802 expression improves glucose tolerance and insulin action. We identify Hnf1b (also known as Tcf2) as a target of miR-802-dependent silencing, and show that short hairpin RNA (shRNA)-mediated reduction of Hnf1b in liver causes glucose intolerance, impairs insulin signalling and promotes hepatic gluconeogenesis. In turn, hepatic overexpression of Hnf1b improves insulin sensitivity in Lepr(db/db) mice. Thus, this study defines a critical role for deregulated expression of miR-802 in the development of obesity-associated impairment of glucose metabolism through targeting of Hnf1b, and assigns Hnf1b an unexpected role in the control of hepatic insulin sensitivity.
|
| [58] |
DOI:10.1073/pnas.1304718110
PMID:23818581
URL
[Cite within: 1]
Placental trophoblasts form the interface between the fetal and maternal environments and serve to limit the maternal-fetal spread of viruses. Here we show that cultured primary human placental trophoblasts are highly resistant to infection by a number of viruses and, importantly, confer this resistance to nonplacental recipient cells by exosome-mediated delivery of specific microRNAs (miRNAs). We show that miRNA members of the chromosome 19 miRNA cluster, which are almost exclusively expressed in the human placenta, are packaged within trophoblast-derived exosomes and attenuate viral replication in recipient cells by the induction of autophagy. Together, our findings identify an unprecedented paracrine and/or systemic function of placental trophoblasts that uses exosome-mediated transfer of a unique set of placental-specific effector miRNAs to directly communicate with placental or maternal target cells and regulate their immunity to viral infections.
|
| [59] |
DOI:10.1038/nm.3287
PMID:3835787
URL
[Cite within: 1]
Abstract Beta-cell dysfunction and impaired insulin production are hallmarks of diabetes, but despite the growing diabetes epidemic, the molecular mechanisms underlying this disease have remained unclear. We identified thioredoxin-interacting protein (TXNIP), a cellular redox regulator, as a crucial factor in beta-cell biology and show that beta-cell TXNIP is upregulated in diabetes, whereas TXNIP deficiency protects against diabetes by preventing beta-cell apoptosis. Here we show that TXNIP and diabetes induce beta-cell expression of a specific microRNA, miR-204, which in turn blocks insulin production by directly targeting and downregulating MAFA, a known insulin transcription factor. In particular, we first discovered the regulation of miR-204 by TXNIP by microarray analysis, followed by validation studies in INS-1 beta cells, islets of Txnip-deficient mice, diabetic mouse models and primary human islets. We then further found that TXNIP induces miR-204 by inhibiting the activity of signal transducer and activator of transcription 3 (STAT3), a transcription factor that is involved in miR-204 regulation. We also identified MAFA as a target that is downregulated by miR-204. Taken together, our results demonstrate that TXNIP controls microRNA expression and insulin production and that miR-204 is involved in beta-cell function. The newly identified TXNIP-miR-204-MAFA-insulin pathway may contribute to diabetes progression and provides new insight into TXNIP function and microRNA biology in health and disease.
|
| [60] |
DOI:10.1073/pnas.1307107110
PMID:23980150
URL
[Cite within: 1]
MicroRNAs (miRNAs) are small 19- to 24-nt noncoding RNAs that have the capacity to regulate fundamental biological processes essential for cancer initiation and progression. In cancer, miRNAs may function as oncogenes or tumor suppressors. Here, we conducted global profiling for miRNAs in a cohort of stage 1 nonsmall cell lung cancers (n = 81) and determined that miR-486 was the most down-regulated miRNA in tumors compared with adjacent uninvolved lung tissues, suggesting that miR-486 loss may be important in lung cancer development. We report that miR-486 directly targets components of insulin growth factor (IGF) signaling including insulin-like growth factor 1 (IGF1), IGF1 receptor (IGF1R), and phosphoinositide-3-kinase, regulatory subunit 1 (alpha) (PIK3R1, or p85a) and functions as a potent tumor suppressor of lung cancer both in vitro and in vivo. Our findings support the role for miR-486 loss in lung cancer and suggest a potential biological link to p53.
|
| [61] |
DOI:10.1016/j.cell.2014.03.055
PMID:24855947
URL
[Cite within: 1]
Abstract Downregulation of the miR-143/145 microRNA (miRNA) cluster has been repeatedly reported in colon cancer and other epithelial tumors. In addition, overexpression of these miRNAs inhibits tumorigenesis, leading to broad consensus that they function as cell-autonomous epithelial tumor suppressors. We generated mice with deletion of miR-143/145 to investigate the functions of these miRNAs in intestinal physiology and disease in vivo. Although intestinal development proceeded normally in the absence of these miRNAs, epithelial regeneration after injury was dramatically impaired. Surprisingly, we found that miR-143/145 are expressed and function exclusively within the mesenchymal compartment of intestine. Defective epithelial regeneration in miR-143/145-deficient mice resulted from the dysfunction of smooth muscle and myofibroblasts and was associated with derepression of the miR-143 target Igfbp5, which impaired IGF signaling after epithelial injury. These results provide important insights into the regulation of epithelial wound healing and argue against a cell-autonomous tumor suppressor role for miR-143/145 in colon cancer. Copyright 2014 Elsevier Inc. All rights reserved.
|
| [62] |
DOI:10.1038/nature11890
PMID:23395958
URL
[Cite within: 1]
Post-transcriptional switches are flexible effectors of dynamic changes in gene expression. Here we report a new post-transcriptional switch that dictates the spatiotemporal and mutually exclusive expression of two alternative gene products from a single transcript. Expression of primate-specific exonic microRNA-198 (miR-198), located in the 30905-untranslated region of follistatin-like 1 (FSTL1) messenger RNA, switches to expression of the linked open reading frame of FSTL1 upon wounding in a human ex vivo organ culture system. We show that binding of a KH-type splicing regulatory protein (KSRP, also known as KHSRP) to the primary transcript determines the fate of the transcript and is essential for the processing of miR-198: transforming growth factor-0205 signalling switches off miR-198 expression by downregulating KSRP, and promotes FSTL1 protein expression. We also show that FSTL1 expression promotes keratinocyte migration, whereas miR-198 expression has the opposite effect by targeting and inhibiting DIAPH1, PLAU and LAMC2. A clear inverse correlation between the expression pattern of FSTL1 (pro-migratory) and miR-198 (anti-migratory) highlights the importance of this regulatory switch in controlling context-specific gene expression to orchestrate wound re-epithelialization. The deleterious effect of failure of this switch is apparent in non-healing chronic diabetic ulcers, in which expression of miR-198 persists, FSTL1 is absent, and keratinocyte migration, re-epithelialization and wound healing all fail to occur.
|
| [63] |
DOI:10.1038/nbt.2666
PMID:3808852
URL
[Cite within: 1]
Recent gain-of-function studies in influenza A virus H5N1 strains revealed that as few as three amino-acid changes in the hemagglutinin protein confer the capacity for viral transmission between ferrets1,2. As transmission between ferrets is considered a surrogate indicator of transmissibility between humans, these studies raised concerns about the risks of gain-of-function influenza A virus research. Here we present an approach to strengthen the biosafety of gain-of-function influenza experiments. We exploit species-specific endogenous small RNAs to restrict influenza A virus tropism. In particular, we found that the microRNA miR-192 was expressed in primary human respiratory tract epithelial cells as well as mouse lungs but absent from the ferret respiratory tract. Incorporation of miR-192 target sites into influenza A virus did not prevent influenza replication and transmissibility in ferrets, but did attenuate influenza pathogenicity in mice. This molecular biocontainment approach should be applicable beyond influenza A virus to minimize the risk of experiments involving other pathogenic viruses.
|
| [64] |
DOI:10.1038/nmeth.2633
PMID:23995387
URL
[Cite within: 1]
Nanoliter-sized droplet technology paired with digital PCR (ddPCR) holds promise for highly precise, absolute nucleic acid quantification. Our comparison of microRNA quantification by ddPCR and real-time PCR revealed greater precision (coefficients of variation decreased 37-86%) and improved day-to-day reproducibility (by a factor of seven) of ddPCR but with comparable sensitivity. When we applied ddPCR to serum microRNA biomarker analysis, this translated to superior diagnostic performance for identifying individuals with cancer.
|
| [65] |
DOI:10.1126/science.1258040
PMID:4313529
URL
[Cite within: 1]
MicroRNAs (miRNAs) control expression of thousands of genes in plants and animals. miRNAs function by guiding Argonaute proteins to complementary sites in messenger RNAs (mRNAs) targeted for repression. We determined crystal structures of human Argonaute-2 (Ago2) bound to a defined guide RNA with and without target RNAs representing miRNA recognition sites. These structures suggest a stepwise mechanism, in which Ago2 primarily exposes guide nucleotides (nt) 2 to 5 for initial target pairing. Pairing to nt 2 to 5 promotes conformational changes that expose nt 2 to 8 and 13 to 16 for further target recognition. Interactions with the guide-target minor groove allow Ago2 to interrogate target RNAs in a sequence-independent manner, whereas an adenosine binding-pocket opposite guide nt 1 further facilitates target recognition. Spurious slicing of miRNA targets is avoided through an inhibitory coordination of one catalytic magnesium ion. These results explain the conserved nucleotide-pairing patterns in animal miRNA target sites first observed over two decades ago.
|
| [66] |
|
| [67] |
DOI:10.1001/jama.2013.284665
PMID:4161014
URL
[Cite within: 1]
Pancreatic cancer represents the 10th most commonly diagnosed cancer, but is the 4th leading cause of cancer-related death in the United States.It is estimated that approximately 4565220 new cases of pancreatic cancer will be diagnosed and that 3865460 people will die of pancreatic cancer in the United States in 2013,with an estimated 22765000 deaths from pancreatic cancer occurring worldwide each year.The incidence of pancreatic cancer has been slowly increasing over the last decade.The 1- and 5-year survival rates for pancreatic cancer are about 25% and 5%, respectively, which are the lowest survival rates of all major cancers.
|
| [68] |
DOI:10.1038/nature13905
PMID:4367962
URL
[Cite within: 1]
Abstract MicroRNAs are short non-coding RNAs expressed in different tissue and cell types that suppress the expression of target genes. As such, microRNAs are critical cogs in numerous biological processes, and dysregulated microRNA expression is correlated with many human diseases. Certain microRNAs, called oncomiRs, play a causal role in the onset and maintenance of cancer when overexpressed. Tumours that depend on these microRNAs are said to display oncomiR addiction. Some of the most effective anticancer therapies target oncogenes such as EGFR and HER2; similarly, inhibition of oncomiRs using antisense oligomers (that is, antimiRs) is an evolving therapeutic strategy. However, the in vivo efficacy of current antimiR technologies is hindered by physiological and cellular barriers to delivery into targeted cells. Here we introduce a novel antimiR delivery platform that targets the acidic tumour microenvironment, evades systemic clearance by the liver, and facilitates cell entry via a non-endocytic pathway. We find that the attachment of peptide nucleic acid antimiRs to a peptide with a low pH-induced transmembrane structure (pHLIP) produces a novel construct that could target the tumour microenvironment, transport antimiRs across plasma membranes under acidic conditions such as those found in solid tumours (pH approximately 6), and effectively inhibit the miR-155 oncomiR in a mouse model of lymphoma. This study introduces a new model for using antimiRs as anti-cancer drugs, which can have broad impacts on the field of targeted drug delivery.
|
| [69] |
DOI:10.1016/j.cell.2013.08.028
PMID:4108076
URL
[Cite within: 1]
Enhancer-associated long noncoding RNAs act over long distances and across chromosomes to activate transcription at distal promoters. Here, we address the latest advances made toward understanding the role of long noncoding RNA expression and the involvement of these RNAs in enhancer function through association with protein factors and modulation of chromatin structure.
|
| [70] |
DOI:10.1038/nature11884
PMID:23417068
URL
[Cite within: 1]
Abstract Recent advances in genomic research have revealed the existence of a large number of transcripts devoid of protein-coding potential in multiple organisms. Although the functional role for long non-coding RNAs (lncRNAs) has been best defined in epigenetic phenomena such as X-chromosome inactivation and imprinting, different classes of lncRNAs may have varied biological functions. We and others have identified a class of lncRNAs, termed ncRNA-activating (ncRNA-a), that function to activate their neighbouring genes using a cis-mediated mechanism. To define the precise mode by which such enhancer-like RNAs function, we depleted factors with known roles in transcriptional activation and assessed their role in RNA-dependent activation. Here we report that depletion of the components of the co-activator complex, Mediator, specifically and potently diminished the ncRNA-induced activation of transcription in a heterologous reporter assay using human HEK293 cells. In vivo, Mediator is recruited to ncRNA-a target genes and regulates their expression. We show that ncRNA-a interact with Mediator to regulate its chromatin localization and kinase activity towards histone H3 serine 10. The Mediator complex harbouring disease- displays diminished ability to associate with activating ncRNAs. Chromosome conformation capture confirmed the presence of DNA looping between the ncRNA-a loci and its targets. Importantly, depletion of Mediator subunits or ncRNA-a reduced the chromatin looping between the two loci. Our results identify the human Mediator complex as the transducer of activating ncRNAs and highlight the importance of Mediator and activating ncRNA association in human disease.
|
| [71] |
DOI:10.1016/j.cell.2013.01.003
PMID:23352431
URL
[Cite within: 1]
Abstract Long noncoding RNAs (lncRNAs) are often expressed in a development-specific manner, yet little is known about their roles in lineage commitment. Here, we identified Braveheart (Bvht), a heart-associated lncRNA in mouse. Using multiple embryonic stem cell (ESC) differentiation strategies, we show that Bvht is required for progression of nascent mesoderm toward a cardiac fate. We find that Bvht is necessary for activation of a core cardiovascular gene network and functions upstream of mesoderm posterior 1 (MesP1), a master regulator of a common multipotent cardiovascular progenitor. We also show that Bvht interacts with SUZ12, a component of polycomb-repressive complex 2 (PRC2), during cardiomyocyte differentiation, suggesting that Bvht mediates epigenetic regulation of cardiac commitment. Finally, we demonstrate a role for Bvht in maintaining cardiac fate in neonatal cardiomyocytes. Together, our work provides evidence for a long noncoding RNA with critical roles in the establishment of the cardiovascular lineage during mammalian development. Copyright 2013 Elsevier Inc. All rights reserved.
|
| [72] |
DOI:10.1126/science.1234848
PMID:5144995
URL
[Cite within: 1]
Abstract Roles for long noncoding RNAs (lncRNAs) in gene expression are emerging, but regulation of the lncRNA itself is poorly understood. We have identified a homeodomain protein, AtNDX, that regulates COOLAIR, a set of antisense transcripts originating from the 3' end of Arabidopsis FLOWERING LOCUS C (FLC). AtNDX associates with single-stranded DNA rather than double-stranded DNA non-sequence-specifically in vitro, and localizes to a heterochromatic region in the COOLAIR promoter in vivo. Single-stranded DNA was detected in vivo as part of an RNA-DNA hybrid, or R-loop, that covers the COOLAIR promoter. R-loop stabilization mediated by AtNDX inhibits COOLAIR transcription, which in turn modifies FLC expression. Differential stabilization of R-loops could be a general mechanism influencing gene expression in many organisms.
|
| [73] |
DOI:10.1016/j.devcel.2013.03.002
PMID:23541921
URL
[Cite within: 1]
Abstract The embryonic stem cell (ESC) transcriptional and epigenetic networks are controlled by a multilayer regulatory circuitry, including core transcription factors (TFs), posttranscriptional modifier microRNAs (miRNAs), and some other regulators. However, the role of large intergenic noncoding RNAs (lincRNAs) in this regulatory circuitry and their underlying mechanism remains undefined. Here, we demonstrate that a lincRNA, linc-RoR, may function as a key competing endogenous RNA to link the network of miRNAs and core TFs, e.g., Oct4, Sox2, and Nanog. We show that linc-RoR shares miRNA-response elements with these core TFs and that linc-RoR prevents these core TFs from miRNA-mediated suppression in self-renewing human ESC. We suggest that linc-RoR forms a feedback loop with core TFs and miRNAs to regulate ESC maintenance and differentiation. These results may provide insights into the functional interactions of the components of genetic networks during development and may lead to new therapies for many diseases. Copyright 2013 Elsevier Inc. All rights reserved.
|
| [74] |
DOI:10.1016/j.devcel.2013.03.020
PMID:23597480
URL
[Cite within: 1]
Large intergenic noncoding (linc) RNAs constitute a new dimension of posttranscriptional gene regulation. In02this issue of Developmental Cell, Wang et02al. (2013) find that linc-RoR maintains human embryonic stem cell self-renewal by functioning as a sponge to trap miR-145, thus regulating core pluripotency factors Oct4, Nanog, and Sox2.
|
| [75] |
DOI:10.1038/nature13671
PMID:25132551
URL
[Cite within: 1]
Abstract Eukaryotic circadian oscillators consist of negative feedback loops that generate endogenous rhythmicities. Natural antisense RNAs are found in a wide range of eukaryotic organisms. Nevertheless, the physiological importance and mode of action of most antisense RNAs are not clear. frequency (frq) encodes a component of the Neurospora core circadian negative feedback loop, which was thought to generate sustained rhythmicity. Transcription of qrf, the long non-coding frq antisense RNA, is induced by light, and its level oscillates in antiphase to frq sense RNA. Here we show that qrf transcription is regulated by both light-dependent and light-independent mechanisms. Light-dependent qrf transcription represses frq expression and regulates clock resetting. Light-independent qrf expression, on the other hand, is required for circadian rhythmicity. frq transcription also inhibits qrf expression and drives the antiphasic rhythm of qrf transcripts. The mutual inhibition of frq and qrf transcription thus forms a double negative feedback loop that is interlocked with the core feedback loop. Genetic and mathematical modelling analyses indicate that such an arrangement is required for robust and sustained circadian rhythmicity. Moreover, our results suggest that antisense transcription inhibits sense expression by mediating chromatin modifications and premature termination of transcription. Taken together, our results establish antisense transcription as an essential feature in a circadian system and shed light on the importance and mechanism of antisense action.
|
| [76] |
DOI:10.1126/science.1251456
PMID:24744378
URL
[Cite within: 1]
Abstract Long noncoding RNAs (lncRNAs) play important roles in diverse biological processes; however, few have been identified that regulate immune cell differentiation and function. Here, we identified lnc-DC, which was exclusively expressed in human conventional dendritic cells (DCs). Knockdown of lnc-DC impaired DC differentiation from human monocytes in vitro and from mouse bone marrow cells in vivo and reduced capacity of DCs to stimulate T cell activation. lnc-DC mediated these effects by activating the transcription factor STAT3 (signal transducer and activator of transcription 3). lnc-DC bound directly to STAT3 in the cytoplasm, which promoted STAT3 phosphorylation on tyrosine-705 by preventing STAT3 binding to and dephosphorylation by SHP1. Our work identifies a lncRNA that regulates DC differentiation and also broadens the known mechanisms of lncRNA action.
|
| [77] |
DOI:10.1016/j.stem.2013.03.003
PMID:3662805
URL
[Cite within: 1]
Genome-wide approaches identifying lncRNAs in subventricular zone neurogenesis provide insight into glial/neuronal lineage specification and are a resource for the study of lncRNAs in development and disease.
|
| [78] |
DOI:10.1038/nature12451
PMID:23945587
URL
[Cite within: 1]
Abstract Although recent studies have indicated roles of long non-coding RNAs (lncRNAs) in physiological aspects of cell-type determination and tissue homeostasis, their potential involvement in regulated gene transcription programs remains rather poorly understood. The androgen receptor regulates a large repertoire of genes central to the identity and behaviour of prostate cancer cells, and functions in a ligand-independent fashion in many prostate cancers when they become hormone refractory after initial androgen deprivation therapy. Here we report that two lncRNAs highly overexpressed in aggressive prostate cancer, PRNCR1 (also known as PCAT8) and PCGEM1, bind successively to the androgen receptor and strongly enhance both ligand-dependent and ligand-independent androgen-receptor-mediated gene activation programs and proliferation in prostate cancer cells. Binding of PRNCR1 to the carboxy-terminally acetylated androgen receptor on enhancers and its association with DOT1L appear to be required for recruitment of the second lncRNA, PCGEM1, to the androgen receptor amino terminus that is methylated by DOT1L. Unexpectedly, recognition of specific protein marks by PCGEM1-recruited pygopus 2 PHD domain enhances selective looping of androgen-receptor-bound enhancers to target gene promoters in these cells. In 'resistant' prostate cancer cells, these overexpressed lncRNAs can interact with, and are required for, the robust activation of both truncated and full-length androgen receptor, causing ligand-independent activation of the androgen receptor transcriptional program and cell proliferation. Conditionally expressed short hairpin RNA targeting these lncRNAs in castration-resistant prostate cancer cell lines strongly suppressed tumour xenograft growth in vivo. Together, these results indicate that these overexpressed lncRNAs can potentially serve as a required component of castration-resistance in prostatic tumours.
|
| [79] |
DOI:10.1242/dev.110544
PMID:25359727
URL
[Cite within: 1]
Abstract Neat1 is a non-protein-coding RNA that serves as an architectural component of the nuclear bodies known as paraspeckles. Although cell-based studies indicate that Neat1 is a crucial regulator of gene expression, its physiological relevance remains unclear. Here, we find that Neat1 knockout (KO) mice stochastically fail to become pregnant despite normal ovulation. Unilateral transplantation of wild-type ovaries or the administration of progesterone partially rescued the phenotype, suggesting that corpus luteum dysfunction and concomitant low progesterone were the primary causes of the decreased fertility. In contrast to the faint expression observed in most of the adult tissues, Neat1 was highly expressed in the corpus luteum, and the formation of luteal tissue was severely impaired in nearly half of the Neat1 KO mice. These observations suggest that Neat1 is essential for the formation of the corpus luteum and for the subsequent establishment of pregnancy under a suboptimal condition that has not yet been identified. 2014. Published by The Company of Biologists Ltd.
|
| [80] |
DOI:10.1073/pnas.1313768111
PMID:24371310
URL
[Cite within: 1]
Abstract Thousands of large intergenic noncoding RNAs (lincRNAs) have been identified in the mammalian genome, many of which have important roles in regulating a variety of biological processes. Here, we used a custom microarray to identify lincRNAs associated with activation of the innate immune response. A panel of 159 lincRNAs was found to be differentially expressed following innate activation of THP1 macrophages. Among them, linc1992 was shown to be expressed in many human tissues and was required for induction of TNF02± expression. Linc1992 bound specifically to heterogenous nuclear ribonucleoprotein L (hnRNPL) and formed a functional linc1992-hnRNPL complex that regulated transcription of the TNF02± gene by binding to its promoter. Transcriptome analysis revealed that linc1992 was required for expression of many immune-response genes, including other cytokines and transcriptional and posttranscriptional regulators of TNF02± expression, and that knockdown of linc1992 caused dysregulation of these genes during innate activation of THP1 macrophages. Therefore, we named linc1992 THRIL (TNF02± and hnRNPL related immunoregulatory LincRNA). Finally, THRIL expression was correlated with the severity of symptoms in patients with Kawasaki disease, an acute inflammatory disease of childhood. Collectively, our data provide evidence that lincRNAs and their binding proteins can regulate TNF02± expression and may play important roles in the innate immune response and inflammatory diseases in humans.
|
| [81] |
DOI:10.1016/j.cell.2014.05.049
PMID:4131209
URL
[Cite within: 1]
Abstract Notch signaling is a key developmental pathway that is subject to frequent genetic and epigenetic perturbations in many different human tumors. Here we investigate whether long noncoding RNA (lncRNA) genes, in addition to mRNAs, are key downstream targets of oncogenic Notch1 in human T0002cell acute lymphoblastic leukemia (T-ALL). By integrating transcriptome profiles with chromatin state maps, we have uncovered many previously unreported T-ALL-specific lncRNA genes, a fraction of which are directly controlled by the Notch1/Rpbj0202 activator complex. Finally we have shown that one specific Notch-regulated lncRNA, LUNAR1, is required for efficient T-ALL growth in0002vitro and in0002vivo due to its ability to enhance IGF1R mRNA expression and sustain IGF1 signaling. These results confirm that lncRNAs are important downstream targets of the Notch signaling pathway, and additionally they are key regulators of the oncogenic state in T-ALL. Copyright 0008 2014 Elsevier Inc. All rights reserved.
|
| [82] |
DOI:10.1038/nature13596
PMID:25119045
URL
[Cite within: 1]
The role of long noncoding RNA (lncRNA) in adult hearts is unknown; also unclear is how lncRNA modulates nucleosome remodeling. An estimated 70% of mouse genes undergo antisense transcription1, including myosin heavy chain 7 (Myh7) that encodes molecular motor proteins for heart contraction2. Here, we identify a cluster of lncRNA transcripts from Myh7 loci and show a new lncRNA–chromatin mechanism for heart failure. In mice, these transcripts, which we named Myosin Heavy Chain Associated RNA Transcripts (MyHEART or Mhrt), are cardiac-specific and abundant in adult hearts. Pathological stress activates the Brg1-Hdac-Parp chromatin repressor complex3 to inhibit Mhrt transcription in the heart. Such stress-induced Mhrt repression is essential for cardiomyopathy to develop: restoring Mhrt to the pre-stress level protects the heart from hypertrophy and failure. Mhrt antagonizes the function of Brg1, a chromatin-remodeling factor that is activated by stress to trigger aberrant gene expression and cardiac myopathy3. Mhrt prevents Brg1 from recognizing its genomic DNA targets, thus inhibiting chromatin targeting and gene regulation by Brg1. Mhrt binds to the helicase domain of Brg1, and this domain is crucial for tethering Brg1 to chromatinized DNA targets. Brg1 helicase has dual nucleic acid-binding specificities: it is capable of binding lncRNA (Mhrt) and chromatinized—but not naked—DNA. This dual-binding feature of helicase enables a competitive inhibition mechanism by which Mhrt sequesters Brg1 from its genomic DNA targets to prevent chromatin remodeling. A Mhrt-Brg1 feedback circuit is thus crucial for heart function. Human MHRT also originates from MYH7 loci and is repressed in various types of myopathic hearts, suggesting a conserved lncRNA mechanism in human cardiomyopathy. Our studies identify the first cardioprotective lncRNA, define a new targeting mechanism for ATP-dependent chromatin-remodeling factors, and establish a new paradigm for lncRNA–chromatin interaction.
|
| [83] |
DOI:10.1016/j.molcel.2013.11.004
PMID:3222224316222
URL
[Cite within: 1]
Abstract Hypoxia has long been linked to the Warburg effect, yet the underlying mechanism remains largely unclear. It is also not known if lncRNAs are involved in the contribution of hypoxia to the Warburg effect. Here we show that lincRNA-p21 is a hypoxia-responsive lncRNA and is essential for hypoxia-enhanced glycolysis. Hypoxia/HIF-102±-induced lincRNA-p21 is able to bind HIF-102± and VHL and thus disrupts the VHL-HIF-102± interaction. This disassociation attenuates VHL-mediated HIF-102± ubiquitination and causes HIF-102± accumulation. These data indicate the existence of a positive feedback loop between HIF-102± and lincRNA-p21 that promotes glycolysis under hypoxia. The ability of lincRNA-p21 to promote tumor growth is validated in mouse xenograft models. Together, these findings suggest that lincRNA-p21 is an important player in the regulation of the Warburg effect and also implicate lincRNA-p21 as a valuable therapeutic target for cancer. Copyright 0008 2014 Elsevier Inc. All rights reserved.
|
| [84] |
|
| [85] |
DOI:10.1126/science.1237973
PMID:3778663
URL
[Cite within: 1]
Many large noncoding RNAs (lncRNAs) regulate chromatin, but the mechanisms by which they localize to genomic targets remain unexplored. We investigated the localization mechanisms of the Xist lncRNA during X-chromosome inactivation (XCI), a paradigm of lncRNA-mediated chromatin regulation. During the maintenance of XCI, Xist binds broadly across the X chromosome. During initiation of XCI, Xist initially transfers to distal regions across the X chromosome that are not defined by specific sequences. Instead, Xist identifies these regions by exploiting the three-dimensional conformation of the X chromosome. Xist requires its silencing domain to spread across actively transcribed regions and thereby access the entire chromosome. These findings suggest a model in which Xist coats the X chromosome by searching in three dimensions, modifying chromosome structure, and spreading to newly accessible locations.
|
| [86] |
|
| [87] |
DOI:10.1038/nature12719
PMID:24162848
URL
[Cite within: 1]
TheXist long noncodingRNA(lncRNA) is essential for X-chromosome inactivation (XCI), the process by which mammals compensate for unequal numbers of sex chromosomes(1-3). During XCI, Xist coats the future inactiveXchromosome(Xi)(4) and recruitsPolycomb repressive complex 2 (PRC2) to the X-inactivation centre (Xic)(5). How Xist spreads silencing on a 150-megabases scale is unclear. Here we generate high-resolution maps of Xist binding on the X chromosome across a developmental time course using CHART-seq. In female cells undergoing XCI de novo, Xist follows a two-step mechanism, initially targeting gene-rich islands before spreading to intervening gene-poor domains. Xist is depleted from genes that escape XCI but may concentrate near escapee boundaries. Xist binding is linearly proportional to PRC2 density and H3 lysine 27 trimethylation (H3K27me3), indicating co-migration of Xist and PRC2. Interestingly, when Xist is acutely stripped off from the Xi in post-XCI cells, Xist recovers quickly within both gene-rich and gene-poor domains on a timescale of hours instead of days, indicating a previously primed Xi chromatin state. We conclude that Xist spreading takes distinct stage-specific forms. During initial establishment, Xist follows a two-step mechanism, but during maintenance, Xist spreads rapidly to both gene-rich and gene-poor regions.
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}